Loss of Proteostasis
;){kind=link}
;){kind=link}
A member of the Hallmarks of Aging [1], the loss of proteostasis is a crucial factor underlying many of the pathological manifestations of aging, particularly neurodegenerative diseases [2]. Recent research has shown that interventions to restore proteostasis, such as through pharmacological agents, genetic modifications, and lifestyle changes, can ameliorate this age-related damage and extend healthy lifespan [3-5].
Definition and mechanisms
Proteostasis ensures that proteins are correctly synthesized, folded, and degraded. This system involves such components as molecular chaperones, the proteasome, and autophagic processes. Together, these pathways form a network that regulates the life cycle of proteins, from synthesis to degradation, ensuring cellular function and survival [6].
Proteins, the building blocks of life, are synthesized by ribosomes, which translate genetic information into functional proteins. Once synthesized, these proteins must fold into their correct three-dimensional structures to perform their biological roles. This process of protein folding is crucial for maintaining cellular function and is an essential aspect of proteostasis [7].
Molecular chaperones are specialized proteins that assist in folding by ensuring proper conformation and preventing aggregation of misfolded proteins [8].
Proteins that are damaged, misfolded, or no longer needed are tagged with ubiquitin and sent to the proteasome for degradation. The ubiquitin-proteasome system (UPS) is essential for eliminating aberrant proteins that can disrupt cellular functions. Autophagy is another important degradation pathway, particularly for larger aggregates and damaged organelles [9].
As organisms age, the mechanisms that ensure proteostasis gradually lose efficiency. This breakdown contributes to the accumulation of damaged or misfolded proteins, a phenomenon observed in several age-related diseases such as Alzheimer’s, Parkinson’s, and Huntington’s diseases [2, 10].
The ability of chaperones and other quality control mechanisms to manage misfolded proteins diminishes with age. As a result, misfolded proteins accumulate, forming aggregates that can interfere with cellular processes [11].
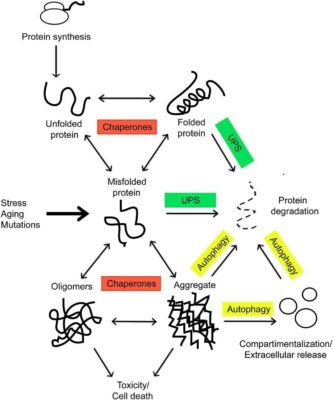
Proteins are produced within the ribosome in an unfolded state. They are met by the chaperones and assisted in folding, which is necessary for a functional protein. Environmental stress, aging, and mutations can tilt the balance toward the formation of misfolded proteins, which tend to form toxic aggregates. These incorrectly folded species are targeted for degradation (Rinaldi et. al, 2016)
Both the ubiquitin-proteasome system and autophagy decline with age. Reduced proteasome activity has been observed in aging tissues, while autophagy, particularly chaperone-mediated autophagy (CMA), becomes less efficient [11].

The accumulation of misfolded or aggregated proteins can be toxic to cells, leading to cellular dysfunction and death. In neurodegenerative diseases, for example, protein aggregates like amyloid-beta plaques and tau tangles are prominent markers of pathology. Furthermore, the failure to clear damaged proteins exacerbates oxidative stress and mitochondrial dysfunction, creating a vicious cycle that accelerates aging [11].
Key pathways
Several key pathways and responses are critical for maintaining proteostasis, including the heat shock response (HSR), the unfolded protein response (UPR), chaperone-mediated autophagy (CMA), and autophagy.
The heat shock response is a protective cellular mechanism activated by stress, particularly elevated temperatures. HSR leads to the increased production of heat shock proteins (HSPs), which help refold damaged proteins or target them for degradation [12].
When misfolded proteins accumulate in the endoplasmic reticulum (ER), the UPR is triggered. This pathway restores normal ER function by halting protein translation, degrading misfolded proteins, and activating the production of molecular chaperones [13].
CMA degrades individual proteins by recognizing a particular motif on the substrate protein and guiding it to lysosomes for degradation. This process becomes less efficient with age, particularly in tissues like the liver, contributing to the buildup of damaged proteins [14].
Autophagy clears larger protein aggregates and damaged organelles, such as defective mitochondria. Autophagosomes form around damaged cellular components and deliver them to lysosomes for degradation. With age, autophagy declines, exacerbating the accumulation of cellular damage [12].
Recent advancements
Research in recent years has provided significant insights into the role of proteostasis in aging and disease. For example, improving the accuracy of ribosomal protein translation can extend lifespan in model organisms. For instance, mutations that reduce error-prone translation in C. elegans and Drosophila enhance longevity by reducing the burden of misfolded proteins [15, 16].
In experimental models, overexpression of molecular chaperones such as HSP70 has been shown to improve proteostasis and extend lifespan by enhancing cells’ ability to refold misfolded proteins or target them for degradation [17].
Compounds such as 4-phenylbutyrate, which reduces ER stress, and proteasome activators have been tested in aging models to promote proteostasis and delay the onset of age-related diseases. In mice, chemical chaperones have been shown to reduce protein aggregation and improve cognitive function [18, 19].
How proteostasis relates to other hallmarks
Proteostasis does not function in isolation; it further contributes to age-related decline through its interaction with other hallmarks of aging. Depending on its state, proteostasis either exacerbates or mitigates aging. Proteostasis is interconnected with other hallmarks of aging, including genomic instability, mitochondrial dysfunction, cellular senescence, disabled macroautophagy, and epigenetic alterations.
Genomic instability
Genomic instability is a hallmark of aging that refers to the accumulation of DNA damage over time. There is a direct relationship between genomic instability and proteostasis. Damaged proteins can impair DNA repair mechanisms [20], while compromised genomic integrity can lead to further dysfunction in proteostasis [21].
When proteostasis fails, the accumulation of misfolded proteins can lead to cellular stress and disrupt the activity of critical enzymes involved in DNA repair. For example, HSPs not only assist in protein folding but also stabilize DNA repair mechanisms. When misfolded proteins overwhelm these systems, cells become less capable of repairing DNA damage, leading to genomic instability [22].
Conversely, genomic instability, which is caused by factors such as oxidative stress, radiation, or replication errors, can impair protein synthesis, potentially leading to more misfolded or incomplete proteins. DNA mutations in genes encoding key proteostasis-related proteins can further deteriorate the protein maintenance network, creating a vicious cycle of dysfunction [23].
Mitochondrial dysfunction
Mitochondrial dysfunction is another critical hallmark of aging that directly influences and is influenced by proteostasis. Mitochondria rely heavily on proteostasis mechanisms to maintain the integrity of their own proteins and metabolic function.
Misfolded proteins within the mitochondria can disrupt the electron transport chain, leading to inefficient energy production and increased production of reactive oxygen species (ROS) [24]. The buildup of damaged mitochondrial proteins, in turn, accelerates mitochondrial dysfunction. Proteostasis mechanisms, including mitochondrial-specific chaperones and proteases, work to clear these defective proteins, but as aging progresses, the capacity of these systems declines [25].
The degradation of dysfunctional mitochondria via autophagy (mitophagy) is a critical process that involves proteostasis. When proteostasis fails, defective mitochondria accumulate, contributing to energy deficits and increased oxidative stress, both of which exacerbate aging [26].
Cellular senescence
Cellular senescence is the irreversible arrest of cell division, often triggered by stress and damage, including the accumulation of protein aggregates [27]. There is a direct link between proteostasis failure and the induction of cellular senescence.
The accumulation of misfolded proteins and aggregates can induce cellular stress responses, leading to a permanent state of senescence. These protein aggregates activate stress pathways, such as the UPR, which, when overwhelmed, activates tumor suppressor pathways, such as p53, causing cells to enter senescence [28].
Senescent cells also experience a decline in proteostasis, further contributing to the dysfunction. These cells accumulate protein aggregates and lose the ability to degrade damaged proteins, exacerbating tissue dysfunction and aging [29].
Disabled macroautophagy
Autophagy, specifically macroautophagy, is vital in maintaining proteostasis by degrading defective proteins and other cellular components. As cells age, macroautophagy becomes increasingly impaired, contributing to the loss of proteostasis [30].
Autophagy is responsible for encapsulating and degrading protein aggregates and damaged organelles. This process is crucial for preventing the buildup of toxic protein aggregates that can impair cellular function. When autophagy is active, it helps sustain proteostasis by continuously removing damaged proteins [31].
With aging, the efficiency of autophagy declines, which leads to the accumulation of damaged proteins and organelles. This decline in autophagy significantly contributes to age-related diseases, such as neurodegenerative disorders in which protein aggregates are a defining feature [32]. Research has shown that enhancing autophagy can restore proteostasis and delay age-related pathologies [33].
Epigenetic alterations
Epigenetic alterations, including changes in DNA methylation and histone modifications, intersect with proteostasis in complex ways. Misfolded proteins can indirectly influence the epigenetic landscape, while proteostasis pathways regulate the activity of epigenetic factors [1].
The accumulation of misfolded proteins can lead to epigenetic changes by activating stress responses that alter the expression of genes related to aging and proteostasis [34]. For instance, the HSR, which is central to proteostasis, is linked to chromatin remodeling and gene expression that protects against cellular stress [5].
Proteostasis pathways, including chaperones and autophagy, play roles in maintaining the integrity of epigenetic machinery. For example, proteins involved in chromatin remodeling and histone modification rely on proper folding and degradation systems. When proteostasis declines, epigenetic regulators may become dysfunctional, contributing to aberrant gene expression patterns associated with aging [35, 36].
Medical advancements and interventions
As proteostasis is a central mechanism in maintaining cellular health and delaying aging, various medical and lifestyle interventions have been developed to preserve or restore proteostasis. These interventions range from pharmacological approaches to gene therapy and lifestyle modifications.
Pharmacological approaches
Pharmacological interventions have emerged as a promising strategy to enhance proteostasis, particularly in neurodegenerative diseases and aging. Candidate drugs include small-molecule chaperones, proteasome activators, and UPR activators.
Small-molecule chaperones are chemical compounds designed to assist in the proper folding of proteins and prevent aggregation. They mimic the action of endogenous chaperones like heat shock proteins, which help proteins to fold. Some of these molecules also stabilize proteins that are prone to misfolding, thus maintaining their function. For instance, Tafamidis, used to treat transthyretin amyloidosis, works by stabilizing the transthyretin protein, preventing its aggregation [8].
Proteasomes are vital in degrading damaged or misfolded proteins as proteins are continuously produced and degraded. Proteasome activity declines with age, contributing to the buildup of protein aggregates. Small molecules like oleuropein and 18α-glycyrrhetinic acid have been shown to activate proteasome activity, improving the clearance of damaged proteins and enhancing cellular proteostasis [37].
While the UPR’s response is crucial in maintaining ER proteostasis and protecting cells from stress, prolonged or excessive UPR activation can lead to cell dysfunction or apoptosis [38]. Pharmacological agents that modulate the UPR, such as Integrated Stress Response Inhibitor (ISRIB), have shown potential in enhancing cognitive function and improving proteostasis, particularly in preclinical models of neurodegeneration [39].
However, the long-term effects of enhancing the UPR remain under investigation, and care must be taken to avoid unintended consequences such as chronic stress or altered cellular processes. Current research suggests that while UPR modulation offers promising therapeutic avenues, its complexity necessitates a careful approach to ensure both efficacy and safety.
Clinical trials
Several clinical trials are underway to explore drugs that can enhance proteostasis and mitigate neurological diseases [40].
Research has focused on compounds that can reduce protein aggregates, such as amyloid plaques and tau tangles. Small molecules like 4-phenylbutyrate (4-PBA), which reduces endoplasmic reticulum stress, are in clinical trials for their ability to improve cognitive function by enhancing proteostasis [18, 19].
Proteostasis-targeting agents like guanabenz, which enhances the UPR and reduces protein misfolding, are being explored in ALS patients. Guanabenz has been shown to delay disease progression by modulating protein degradation pathways [41].
These clinical trials are significant because they aim to manage symptoms and address the underlying mechanisms of proteostasis collapse in neurodegenerative diseases.
Gene therapy
Gene therapy holds significant promise for improving proteostasis by enhancing autophagy. One promising approach involves the overexpression of Lysosome-Associated Membrane Protein 2A (LAMP2A), a key regulator of chaperone-mediated autophagy (CMA). CMA selectively degrades specific proteins, and research in mouse models has demonstrated that increasing LAMP2A expression improves CMA activity, enhancing proteostasis and reducing the accumulation of age-related protein aggregates [42].
This strategy has been shown to restore proteostasis in organs such as the liver, and there is potential for expanding this approach to treat neurodegenerative diseases, where protein aggregation is a hallmark. By targeting LAMP2A and other elements of the autophagic system, gene therapy offers a way to address proteostasis decline, particularly in age-affected tissues like the brain and liver [43].
Diet and lifestyle interventions
In addition to pharmacological and gene therapy interventions, lifestyle changes such as caloric restriction, intermittent fasting, and ketogenic diets have been shown to impact proteostasis and promote longevity profoundly [4].
Reducing calorie intake without malnutrition has been consistently shown to extend lifespan in various organisms, from yeast to mammals. CR enhances proteostasis by upregulating autophagy, reducing protein synthesis, and improving the removal of damaged proteins. This process has been linked to suppressing the mTOR pathway, which promotes autophagy and proteasomal activity [44, 45]. Studies suggest intermittent fasting can activate pathways similar to CR.
The ketogenic diet, which is low in carbohydrates and high in fats, induces the production of ketone bodies, particularly β-hydroxybutyrate (BHB). BHB has been shown to enhance mitochondrial function and promote proteostasis by activating autophagy and reducing oxidative damage to proteins [46-48].
Epigenetic clocks
Based on DNA methylation patterns, epigenetic clocks have emerged as powerful tools to predict biological age and evaluate the effectiveness of anti-aging interventions. Proteostasis-modifying treatments are now being explored for their potential to reset or slow down these epigenetic clocks.
As the aging process progresses, epigenetic changes accumulate, affecting the expression of genes involved in proteostasis. Emerging research suggests that restoring proteostasis through pharmacological, genetic, or lifestyle interventions can influence epigenetic clocks and potentially reverse aspects of aging [49].
Specific proteostasis-modifying treatments, such as caloric restriction and autophagy inducers, can slow down or even reverse epigenetic aging. For example, a study demonstrated that treatment with a combination of growth hormone, DHEA, and metformin could reverse the epigenetic clock in humans, with proteostasis likely playing a key role in this rejuvenation [50].
Research is ongoing to determine whether direct interventions in proteostasis could be used as part of a broader anti-aging strategy to modulate epigenetic clocks. As proteostasis is a fundamental process tied to the regulation of aging, treatments aimed at maintaining protein homeostasis will likely play a crucial role in future anti-aging therapies [51].
Integrated approaches
As interconnected biological processes drive aging, future research must focus on integrated approaches that target multiple hallmarks simultaneously. Proteostasis, while central, does not act in isolation; its dysfunction accelerates other age-related processes, such as mitochondrial dysfunction, cellular senescence, and chronic inflammation. There is a growing interest in combining proteostasis-based therapies with other anti-aging interventions:
Senolytics selectively clear senescent cells and could work synergistically with proteostasis-enhancing therapies. Senescent cells accumulate misfolded proteins and disrupt tissue homeostasis. Removing these cells while simultaneously improving proteostasis could significantly enhance tissue function and slow aging.
Mitochondrial dysfunction is closely linked to impaired proteostasis, as damaged mitochondria accumulate misfolded proteins and oxidative stress. Combining mitochondrial-targeting drugs like NAD+ precursors or mitochondrial uncouplers with proteostasis enhancers could optimize cellular energy production and protein homeostasis, leading to improved healthspan.
Chronic, low-grade inflammation (inflammaging), a hallmark of aging, is often triggered by the accumulation of damaged proteins and dysfunctional organelles. When combined with proteostasis-targeting treatments such as autophagy enhancers, anti-inflammatory therapies could mitigate the pro-inflammatory effects of protein aggregates, improving overall cellular health.
Challenges and opportunities
While proteostasis-targeting therapies offer great promise, translating these findings from animal models to human clinical applications remains a significant challenge.
Many proteostasis-enhancing interventions, such as genetic modifications of autophagy-related genes, have shown profound effects in animal models. However, translating these interventions to humans is complex due to differences in biology and lifespan. Human trials must account for long-term safety and efficacy, especially in aging.
Another challenge is determining the optimal dosage, timing, and combination of proteostasis-enhancing therapies. Proteostasis is a dynamic process, and excessive or poorly timed interventions could disrupt the delicate balance of protein synthesis and degradation, potentially leading to unintended side effects.
Identifying reliable biomarkers to track improvements in proteostasis is crucial for the clinical success of these therapies. While progress has been made with epigenetic clocks and protein aggregate measurements, more research is needed to establish comprehensive markers of proteostasis that can guide therapeutic interventions in humans.
Despite these challenges, the opportunities are vast. As our understanding of proteostasis deepens, it becomes clearer that these therapies hold the potential to significantly impact human aging and healthspan.
Potential of proteostasis-based therapies
Proteostasis-based therapies could fundamentally reshape the future of aging by targeting one of the core mechanisms driving age-related diseases. If successfully translated to human applications, the following effects could be anticipated:
By maintaining protein homeostasis across tissues, these therapies could improve tissue function and resilience, leading to extended healthspan. This would delay the onset of multiple age-related diseases, improving quality of life and longevity.
Emerging research suggests that proteostasis-enhancing interventions may be able to reset aspects of biological age, as indicated by epigenetic clocks. This could lead to therapies extending lifespan and reversing age-related decline, offering new hope for regenerative medicine.
With the rapid advancement in biotechnology, the next few decades could witness significant breakthroughs in proteostasis research, leading to therapies that could revolutionize the way we approach aging and age-related diseases.
Conclusion
Proteostasis plays a crucial role in aging by maintaining the balance between protein synthesis, folding, and degradation. As we age, the loss of proteostasis leads to the accumulation of misfolded proteins, contributing to neurodegenerative diseases, cellular dysfunction, and tissue degeneration. The collapse of proteostasis is linked to other hallmarks of aging, such as mitochondrial dysfunction, cellular senescence, and epigenetic alterations, underscoring its importance as a central mechanism in aging.
Recent medical advances, including small molecule chaperones, proteasome activators, and gene therapy targeting proteostasis, have shown promising results in extending healthspan and delaying age-related diseases. Lifestyle interventions such as caloric restriction and ketogenic diets have also demonstrated the ability to enhance proteostasis and promote longevity. These developments suggest that maintaining proteostasis will be critical in future strategies to mitigate aging and improve quality of life.
However, continued research is needed to unlock the potential of proteostasis-based therapies fully. Integrating proteostasis-enhancing interventions with treatments targeting other hallmarks of aging, such as senolytics and mitochondrial enhancers, could offer synergistic benefits. Moreover, the challenge of translating these therapies from animal models to humans must be addressed through rigorous clinical trials and the development of reliable biomarkers.
In conclusion, improving proteostasis represents a promising avenue for extending health span and potentially reshaping human aging. As the field continues to evolve, it will be essential to develop integrated, multi-targeted strategies that address the complexity of aging and harness the power of proteostasis for longevity.
Literature
[1] López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278..
[2] Panwar, S.; Uniyal, P.; Kukreti, N.; Hashmi, A.; Verma, S.; Arya, A.; Joshi, G. Role of Autophagy and Proteostasis in Neurodegenerative Diseases: Exploring the Therapeutic Interventions. Chem Biol Drug Des 2024, 103.
[3] Lim, J.J.; Noh, S.; Kang, W.; Hyun, B.; Lee, B.H.; Hyun, S. Pharmacological Inhibition of USP14 Delays Proteostasis-Associated Aging in a Proteasome-Dependent but Foxo-Independent Manner. Autophagy 2024.
[4] Almeida, M.F.; Farizatto, K.L.G.; Almeida, R.S.; Bahr, B.A. Lifestyle Strategies to Promote Proteostasis and Reduce the Risk of Alzheimer’s Disease and Other Proteinopathies. Ageing Res Rev 2024, 93, 102162.
[5] Pessa, J.C.; Joutsen, J.; Sistonen, L. Transcriptional Reprogramming at the Intersection of the Heat Shock Response and Proteostasis. Mol Cell 2024, 84, 80–93.
[6] Morimoto, R.I.; Ktistakis, N.T. Proteostasis in Health and Disease: A Conversation with Professor Rick Morimoto. Autophagy 2024.
[7] Green, R.; Noller, H.F. Ribosomes and Translation. Annu Rev Biochem 1997, 66, 679–716.
[8] Sharma, A.; Shah, O.P.; Sharma, L.; Gulati, M.; Behl, T.; Khalid, A.; Mohan, S.; Najmi, A.; Zoghebi, K. Molecular Chaperones as Therapeutic Target: Hallmark of Neurodegenerative Disorders. Molecular Neurobiology 2023 61:7 2023, 61, 4750–4767.
[9] Upadhyay, A.; Joshi, V. The Ubiquitin Tale: Current Strategies and Future Challenges. ACS Pharmacol Transl Sci 2024.
[10] Abramovich, J.; Kleczko, K.; Masto, V.; Frydman, J. Proteostasis Function and Dysfunction in Huntington’s Disease. Huntington’s Disease: Pathogenic Mechanisms and Implications for Therapeutics 2024, 205–227.
[11] Hipp, M.S.; Kasturi, P.; Hartl, F.U. The Proteostasis Network and Its Decline in Ageing. Nat Rev Mol Cell Biol 2019, 20, 421–435.
[12] Singh, M.K.; Shin, Y.; Ju, S.; Han, S.; Choe, W.; Yoon, K.S.; Kim, S.S.; Kang, I. Heat Shock Response and Heat Shock Proteins: Current Understanding and Future Opportunities in Human Diseases. International Journal of Molecular Sciences 2024, Vol. 25, Page 4209 2024, 25, 4209.
[13] Kim, P. Understanding the Unfolded Protein Response (UPR) Pathway: Insights into Neuropsychiatric Disorders and Therapeutic Potentials. Biomol Ther (Seoul) 2024, 32, 183.
[14] Yamamoto, H.; Matsui, T. Molecular Mechanisms of Macroautophagy, Microautophagy, and Chaperone-Mediated Autophagy. Journal of Nippon Medical School 2024, 91, 2–9.
[15] Ke, Z.; Mallik, P.; Johnson, A.B.; Luna, F.; Nevo, E.; Zhang, Z.D.; Gladyshev, V.N.; Seluanov, A.; Gorbunova, V. Translation Fidelity Coevolves with Longevity. Aging Cell 2017, 16, 988–993.
[16] Martinez-Miguel, V.E.; Lujan, C.; Espie–Caullet, T.; Martinez-Martinez, D.; Moore, S.; Backes, C.; Gonzalez, S.; Galimov, E.R.; Brown, A.E.X.; Halic, M.; et al. Increased Fidelity of Protein Synthesis Extends Lifespan. Cell Metab 2021, 33, 2288-2300.e12.
[17] Bobkova, N. V.; Evgen’ev, M.; Garbuz, D.G.; Kulikov, A.M.; Morozov, A.; Samokhin, A.; Velmeshev, D.; Medvinskaya, N.; Nesterova, I.; Pollock, A.; et al. Exogenous Hsp70 Delays Senescence and Improves Cognitive Function in Aging Mice. Proc Natl Acad Sci U S A 2015, 112, 16006–16011.
[18] Hafycz, J.M.; Strus, E.; Naidoo, N. Reducing ER Stress with Chaperone Therapy Reverses Sleep Fragmentation and Cognitive Decline in Aged Mice. Aging Cell 2022, 21, e13598.
[19] Yu, Q.; Wang, Z.; Tu, Y.; Cao, Y.; Zhu, H.; Shao, J.; Zhuang, R.; Zhou, Y.; Zhang, J. Proteasome Activation: A Novel Strategy for Targeting Undruggable Intrinsically Disordered Proteins. Bioorg Chem 2024, 145, 107217.
[20] González-Quiroz, M.; Blondel, A.; Sagredo, A.; Hetz, C.; Chevet, E.; Pedeux, R. When Endoplasmic Reticulum Proteostasis Meets the DNA Damage Response. Trends Cell Biol 2020, 30, 881–891.
[21] Lehmann, C.P.; González-Fernández, P.; Tercero, J.A. Spatial Regulation of DNA Damage Tolerance Protein Rad5 Interconnects Genome Stability Maintenance and Proteostasis Networks. Nucleic Acids Res 2024, 52, 1156–1172.
[22] Wen, J.H.; He, X.H.; Feng, Z. Sen; Li, D.Y.; Tang, J.X.; Liu, H.F. Cellular Protein Aggregates: Formation, Biological Effects, and Ways of Elimination. International Journal of Molecular Sciences 2023, Vol. 24, Page 8593 2023, 24, 8593.
[23] Huiting, W.; Bergink, · Steven Locked in a Vicious Cycle: The Connection between Genomic Instability and a Loss of Protein Homeostasis. Genome Instability & Disease 2020 2:1 2020, 2, 1–23.
[24] Zarges, C.; Riemer, J. Oxidative Protein Folding in the Intermembrane Space of Human Mitochondria. FEBS Open Bio 2024.
[25] Baker, B.M.; Haynes, C.M. Mitochondrial Protein Quality Control during Biogenesis and Aging. Trends Biochem Sci 2011, 36, 254–261.
[26] Drake, J.C.; Yan, Z. Mitophagy in Maintaining Skeletal Muscle Mitochondrial Proteostasis and Metabolic Health with Ageing. J Physiol 2017, 595, 6391–6399.
[27] Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annual Review of Pathology: Mechanisms of Disease 2010, 5, 99–118.
[28] Thapa, R.; Ahmad Bhat, A.; Shahwan, M.; Ali, H.; PadmaPriya, G.; Bansal, P.; Rajotiya, S.; Barwal, A.; Siva Prasad, G. V.; Pramanik, A.; et al. Proteostasis Disruption and Senescence in Alzheimer’s Disease Pathways to Neurodegeneration. Brain Res 2024, 1845, 149202.
[29] Sabath, N.; Levy-Adam, F.; Younis, A.; Rozales, K.; Meller, A.; Hadar, S.; Soueid-Baumgarten, S.; Shalgi, R. Cellular Proteostasis Decline in Human Senescence. Proc Natl Acad Sci U S A 2020, 117, 31902–31913
[30] Li, J.; Moretti, F.; Hidvegi, T.; Sviben, S.; Fitzpatrick, J.A.J.; Sundaramoorthi, H.; Pak, S.C.; Silverman, G.A.; Knapp, B.; Filipuzzi, I.; et al. Multiple Genes Core to ERAD, Macroautophagy and Lysosomal Degradation Pathways Participate in the Proteostasis Response in Α1-Antitrypsin Deficiency. Cell Mol Gastroenterol Hepatol 2024, 17, 1007–1024.
[31] Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741.
[32] Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and Aging: The Importance of Maintaining “Clean” Cells. Autophagy 2005, 1, 131–140.
[33] Moors, T.E.; Hoozemans, J.J.M.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.C.; Van De Berg, W.D.J. Therapeutic Potential of Autophagy-Enhancing Agents in Parkinson’s Disease. Molecular Neurodegeneration 2017 12:1 2017, 12, 1–18.
[34] D’Amico, D.; Sorrentino, V.; Auwerx, J. Cytosolic Proteostasis Networks of the Mitochondrial Stress Response. Trends Biochem Sci 2017, 42, 712–725.
[35] Cugusi, S.; Mitter, R.; Kelly, G.P.; Walker, J.; Han, Z.; Pisano, P.; Wierer, M.; Stewart, A.; Svejstrup, J.Q. Heat Shock Induces Premature Transcript Termination and Reconfigures the Human Transcriptome. Mol. Cell 2022, 82, 1573-1588.e10.
[36] Alagar Boopathy, L.R.; Jacob-Tomas, S.; Alecki, C.; Vera, M. Mechanisms Tailoring the Expression of Heat Shock Proteins to Proteostasis Challenges. Journal of Biological Chemistry 2022, 298, 101796
[37] Rao, N.R.; Upadhyay, A.; Savas, J.N. Derailed Protein Turnover in the Aging Mammalian Brain. Mol Syst Biol 2024, 20, 120–139.
[38] Bester, D.; Blignaut, M.; Huisamen, B. ATM Facilitates Autophagy and Protects against Oxidative Stress and Apoptosis in Response to ER Stress in Vitro. Biochem Biophys Res Commun 2024, 732.
[39] Chou, A.; Krukowski, K.; Jopson, T.; Zhu, P.J.; Costa-Mattioli, M.; Walter, P.; Rosi, S. Inhibition of the Integrated Stress Response Reverses Cognitive Deficits after Traumatic Brain Injury. Proc Natl Acad Sci U S A 2017, 114, E6420–E6426.
[40] Panwar, S.; Uniyal, P.; Kukreti, N.; Hashmi, A.; Verma, S.; Arya, A.; Joshi, G. Role of Autophagy and Proteostasis in Neurodegenerative Diseases: Exploring the Therapeutic Interventions. Chem Biol Drug Des 2024, 103, e14515.
[41] Dalla Bella, E.; Bersano, E.; Antonini, G.; Borghero, G.; Capasso, M.; Caponnetto, C.; Chiò, A.; Corbo, M.; Filosto, M.; Giannini, F.; et al. The Unfolded Protein Response in Amyotrophic Later Sclerosis: Results of a Phase 2 Trial. Brain 2021, 144, 2635–2647
[42] Li, X.; Meng, Z.; He, X.; Wang, J.; Yang, L.; Fang, Z. Resveratrol Attenuates Neuroinflammation and Alleviates Emotional Dysfunction in Mice with Sepsis-Associated Encephalopathy through Promoting Chaperone-Mediated Autophagy (CMA). Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2024, 40, 481–487.
[43] Endicott, S.J.; Boynton, D.N.; Beckmann, L.J.; Miller, R.A. Long-Lived Mice with Reduced Growth Hormone Signaling Have a Constitutive Upregulation of Hepatic Chaperone-Mediated Autophagy. Autophagy 2021, 17, 612–625.
[44] Zhang, R.; Wang, X.; Qu, J.H.; Liu, B.; Zhang, P.; Zhang, T.; Fan, P.C.; Wang, X.M.; Xiao, G.Y.; Su, Y.; et al. Caloric Restriction Induces MicroRNAs to Improve Mitochondrial Proteostasis. iScience 2019, 17, 155–166.
[45] Gupta, N.J. Fasting Helps Nutrient Sensing Systems in Clocking the Metabolism. Aging Pathobiol Ther 2024, 0, 60–66.
[46] Munroe, M.; Mahmassani, Z.S.; Dvoretskiy, S.; Reid, J.J.; Miller, B.F.; Hamilton, K.; Rhodes, J.S.; Boppart, M.D. Cognitive Function Is Preserved in Aged Mice Following Long-Term β-Hydroxy β-Methylbutyrate Supplementation. Nutr Neurosci 2020, 23, 170–182.
[47] Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem Res 2017, 42, 35–49.
[48] García-Velázquez, L.; Massieu, L. The Proteomic Effects of Ketone Bodies: Implications for Proteostasis and Brain Proteinopathies. Front Mol Neurosci 2023, 16, 1214092.
[49] Jiang, S.; Guo, Y. Epigenetic Clock: DNA Methylation in Aging. Stem Cells Int 2020, 2020, 1047896.
[50] Fahy, G.M.; Brooke, R.T.; Watson, J.P.; Good, Z.; Vasanawala, S.S.; Maecker, H.; Leipold, M.D.; Lin, D.T.S.; Kobor, M.S.; Horvath, S. Reversal of Epigenetic Aging and Immunosenescent Trends in Humans. Aging Cell 2019, 18, e13028
[51] Thompson, M.A.; De-Souza, E.A. A Year at the Forefront: A Year at the Forefront of Proteostasis and Aging. Biol Open 2023, 12.