Boosting a key autophagy-related protein discourages a core component of Alzheimer’s from taking hold, according to a study published in Aging Cell.
Taking out the trash
Autophagy is the maintenance process of the cell, in which autophagosomes engulf unwanted organelles and other material and fuse together with lysosomes to be digested. As these unwanted components include such things as misfolded proteins, this is far from the first study to link autophagic deficiencies to Alzheimer’s [1, 2].
Along with the well-known amyloid beta, misfolded and modified tau is the key biomarker of Alzheimer’s disease. Tau is a necessary protein for brain function, as it provides key functions for structure and signaling [3]; however, it can also be modified in a very large number of ways, many of which lead to the death of neurons and thus cognitive decline [4]. The most well-known, and possibly most dangerous, is phosphorylation, and phosphorylated tau has been known to be core to Alzheimer’s for decades [5]. Even worse, an excess of misfolded tau can cause failures in autophagy, leading to a rapid increase in the related problems [6].
To fight back against this process and restore autophagy to distressed neurons, this research focuses on tectonin beta-propeller repeat-containing protein 1 (TECPR1), which encourages autophagosomes and lysosomes to fuse [7], accelerates the consumption of protein aggregates, including in stem cells [8], and repairs damaged lysosomes [9]. However, TECPR1 had never been previously investigated in the context of Alzheimer’s.
Tau tangles lead to impaired clearance
This study began by causing a harmful, mutated form of tau, P301S-tau, to form in HEK293 human kidney cells. They found two harmful effects: first, that P301S-tau was discouraging autophagosomes from forming in the first place and then that this form of tau was preventing autophagosomes and lysosomes from combining.
This finding was replicated in mice. Transgenic mice that expressed P301S-tau actually had more autophagosomes than wild-type mice; they were just unable to complete their jobs, being left free-floating within the cell. As the researchers expected, there was far less TECPR1 in the cells of the transgenic mice, including in hippocampal neurons, which are responsible for learning and memory; this held true whether the mice were born transgenic or transfected with a retrovirus at a young age. The levels of other autophagy-related proteins were also heavily dysregulated.
Transfecting HEK293 cells with TECPR1 appeared to do the opposite of P301S-tau. More autophagosomes were created in the TECPR1-transfected cells, and autosomal and lysosomal fusion was increased as well.
TECPR1 fights tau tangles in mice
With these positive results in hand, the researchers then turned to their mouse population. 8-month-old wild-type and P301S mice were transfected with a retrovirus that causes the overexpression of TECPR1, then studied a month later. In wild-type mice, this did nothing in terms of brain capability; there were no changes in learning ability nor behavior.
However, in the P301S group, there were a few marked changes. In the Morris water maze test, P301S mice were much slower to explore, and their memory was much worse. Transfection with TECPR1 brought these metrics much closer to those of wild-type mice. The transfection also caused benefits in object recognition; TECPR1-treated P301S mice were much better at distinguishing between new and old objects than their untreated, tau-tangled counterparts, and they had a greater ability to retain fear memories as well.
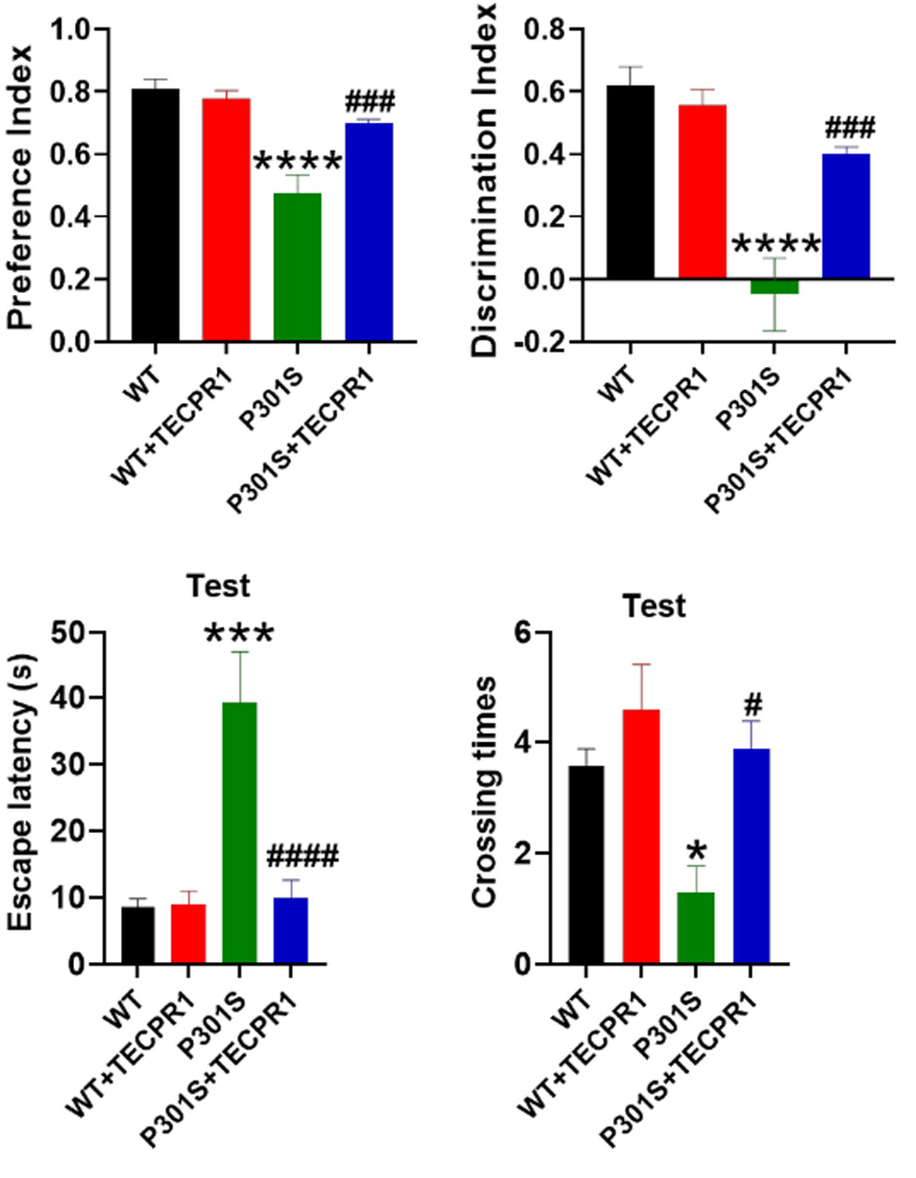
These findings were confirmed when the mice’s brains were analyzed. While TECPR1 did nothing beneficial for wild-type mice, using it to combat the mutant tau caused the neurons to stay alive and to make more connections with other neurons. Fundamental proteins that were reduced with P301S were restored with TECPR1. Overall, the researchers concluded that TECPR1 restores neuroplasticity to tau-impaired mice.
Further work found that the mechanism of action was indeed as the researchers had believed: both total tau and phosphorylated tau were reduced in the hippocampi of the P301S mice. An examination of gene expression found that TECPR1 did not affect the production of tau, only its consumption, and further work found that administering other autophagy-inhibiting compounds will prevent TECPR1 from having any positive effect.
With these results, the researchers believe that TECPR1 is a good target for treating Alzheimer’s disease. However, there are no known methods of getting more TECPR1 into the neurons of living people. To begin a clinical trial, either a gene therapy, ideally one that only targets the affected neurons, must be developed or a small molecule or nanoparticle must be found to efficiently administer TECPR1 into the affected cells or to cause them to upregulate it themselves.
Literature
[1] Zhang, Z., Yang, X., Song, Y. Q., & Tu, J. (2021). Autophagy in Alzheimer’s disease pathogenesis: Therapeutic potential and future perspectives. Ageing research reviews, 72, 101464.
[2] Zhang, W., Xu, C., Sun, J., Shen, H. M., Wang, J., & Yang, C. (2022). Impairment of the autophagy–lysosomal pathway in Alzheimer’s diseases: pathogenic mechanisms and therapeutic potential. Acta Pharmaceutica Sinica B, 12(3), 1019-1040.
[3] Wang, J. Z., & Liu, F. (2008). Microtubule-associated protein tau in development, degeneration and protection of neurons. Progress in neurobiology, 85(2), 148-175.
[4] Li, C., & Götz, J. (2017). Tau-based therapies in neurodegeneration: opportunities and challenges. Nature Reviews Drug Discovery, 16(12), 863-883.
[5] Braak, H., & Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta neuropathologica, 82(4), 239-259.
[6] Feng, Q., Luo, Y., Zhang, X. N., Yang, X. F., Hong, X. Y., Sun, D. S., … & Wang, J. Z. (2020). MAPT/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: a vicious cycle in Alzheimer neurodegeneration. Autophagy, 16(4), 641-658.
[7] Kim, J. H., Hong, S. B., Lee, J. K., Han, S., Roh, K. H., Lee, K. E., … & Song, H. K. (2015). Insights into autophagosome maturation revealed by the structures of ATG5 with its interacting partners. Autophagy, 11(1), 75-87.
[8] Wetzel, L., Blanchard, S., Rama, S., Beier, V., Kaufmann, A., & Wollert, T. (2020). TECPR1 promotes aggrephagy by direct recruitment of LC3C autophagosomes to lysosomes. Nature communications, 11(1), 2993.
[9] Corkery, D. P., Castro‐Gonzalez, S., Knyazeva, A., Herzog, L. K., & Wu, Y. W. (2023). An ATG12‐ATG5‐TECPR1 E3‐like complex regulates unconventional LC3 lipidation at damaged lysosomes. EMBO reports, 24(9), e56841.