Scientists have discovered that ATP deficiency disrupts dopamine processing in synapses, leading to the accumulation of the harmful protein species that characterize Parkinson’s disease. ATP supplementation helps, but the road to the clinic might be long [1].
Parkinson’s and dopamine
Parkinson’s disease is defined by two hallmarks: the death of dopamine-producing neurons in a midbrain region called the substantia nigra, and the accumulation of the protein α-synuclein into clumps known as Lewy bodies [2]. Dopamine is a neurotransmitter, a chemical messenger that neurons use to communicate, and its loss has been linked to Parkinson’s symptoms such as tremor, rigidity, and movement difficulties [3].
Inside neurons, dopamine, being chemically unstable, must be rapidly packaged into membrane-enclosed bubbles called synaptic vesicles to prevent it from oxidizing, as its oxidized form can damage proteins and organelles. The key molecule responsible for loading dopamine into vesicles is vesicular monoamine transporter 2 (VMAT2), which functions as a pump embedded in the vesicle membrane that actively pushes dopamine inside. Crucially, this pump requires energy in the form of ATP, the cell’s universal energy currency, supplied by resident mitochondria.
DJ-1 is a small protein involved in mitochondrial quality control and cellular antioxidant defense. Mutations in the PARK7 gene, which codes for DJ-1, cause a rare, early-onset form of familial Parkinson’s. Importantly, prior studies have hinted that DJ-1 can directly stimulate VMAT2, but this had never been investigated in genuine human dopaminergic neurons. Scientists from Ludwig Maximilian University of Munich attempted to bridge this knowledge gap in this new study published in Science Advances. The same team had previously shown that this mechanism is probably also relevant for sporadic Parkinson’s, which constitutes a majority of cases [4].
Interestingly, mouse models of Parkinson’s have historically failed to reproduce the dopamine oxidation and neuron death characteristic of human disease [4]. The authors, however, used neurons derived from human induced pluripotent stem cells (iPSCs), a model far more disease-relevant than rodent systems for this particular question.
Connecting the dots
After differentiating iPSCs from two DJ-1 knockout lines and their paired healthy controls into mature midbrain dopaminergic neurons, the researchers performed a proteomics analysis and found that VMAT2 was among the most downregulated proteins in the DJ-1-KO lines. Gene ontology analysis of the downregulated proteins implicated pathways related to synaptic vesicles, transmembrane transporter activity, and chemical synaptic transmission.
Confocal microscopy showed reduced density of dopaminergic VMAT2-positive synapses in DJ-1 KO neurons. Using an even more advanced imaging technology, MINFLUX-DNA PAINT, an ultra-high-resolution single-molecule imaging method, the researchers were able to look at individual dopaminergic synapses. Both the total number of VMAT2-positive vesicles per synapse and the copy number of VMAT2 molecules per vesicle were reduced; the latter directly determines how much dopamine can be loaded into each vesicle.
Next, the scientists added a third model: neurons derived from an actual patient with DJ-1-linked Parkinson’s. VMAT2 protein levels, ATP levels, and mitochondrial membrane potentials were significantly reduced in all three DJ-1-deficient lines. This suggests that the vesicle-loading defect is not just anatomical but functional: the neurons do not merely have less VMAT2 but actually sequester their cargo less effectively, and ATP deficit is directly relevant to that effect.
Vesicles also showed morphological abnormalities. Researchers were able to connect this to clathrin-mediated vesicle recycling: clathrin is the protein that covers newly endocytosed vesicles and needs to be removed in a process that uses ATP, just like dopamine loading. If ATP is low, vesicle uncoating could stall, producing malformed vesicles. Consistent with that, clathrin levels were roughly doubled in DJ-1-deficient neurons.
In the crucial next experiment, the team confirmed that DJ-1-deficient neurons had elevated oxidized dopamine and more of both total α-synuclein and its pathological species. This finalized a clear chain of events: vesicular dopamine handling breaks down, dopamine oxidizes, and pathological α-synuclein rises.

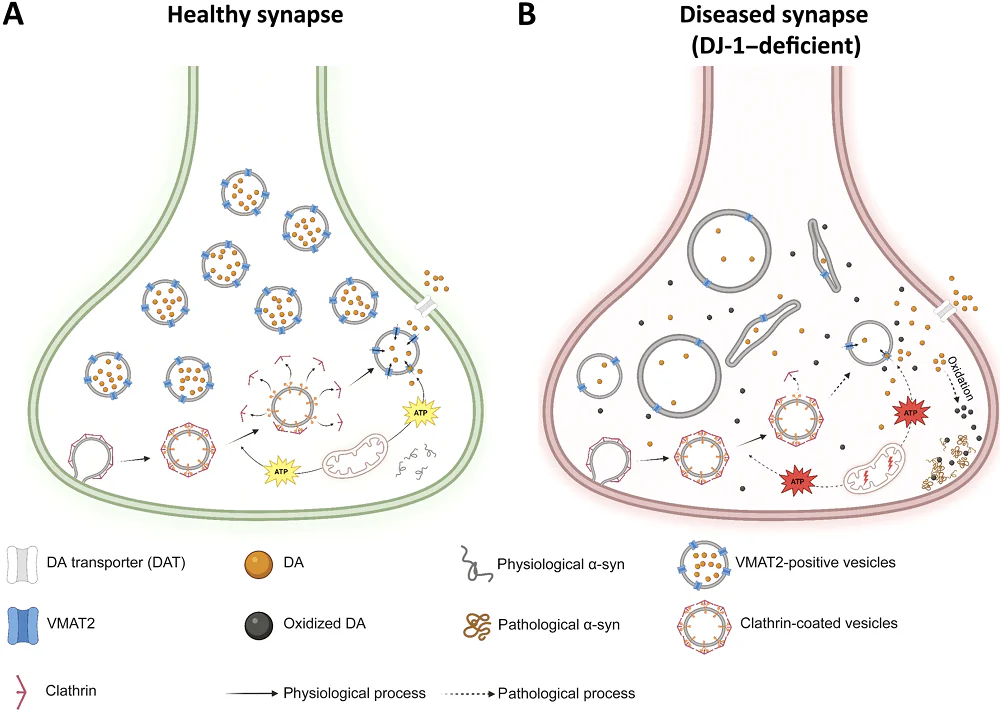
ATP rescues the pathology
Finally, the team ran a rescue experiment, simply treating DJ-1-deficient neurons with ATP for 72 hours and using a “broken” ATP analog as control. As a result, vesicular loading went up while the levels of clathrin, oxidized dopamine, and total and pathological α-synuclein fell.
“Dopamine oxidizes to produce toxic substances and causes lasting damage to the neurons if it is not properly packaged in vesicles – but the cause of this dysfunctional packaging of dopamine was hitherto unclear,” said Lena Burbulla, Professor of Metabolic Biochemistry in the Faculty of Medicine at LMU and a senior author of the study. “The lack of DJ-1 causes energy problems that occur in many variants of Parkinson’s. This discovery links energy deficiency to the packaging of dopamine and neuron vulnerability – a new mechanism for Parkinson’s.”
Interestingly, the team also ran a set of experiments with mouse DJ-1 knockout iPSC-derived dopaminergic neurons, which also showed reduced VMAT2 protein but did not accumulate oxidized dopamine. The authors hypothesize that rodents may have compensatory dopamine-handling mechanisms that humans lack or use less effectively. This both highlights the limitations of murine PD models and suggests that these rodent-specific mechanisms might potentially be studied and translated into humans.
While this proof-of-concept in vitro study suggests a possible rescue route for PD, the way to the clinic might be long and bumpy, as effective ATP supplementation, especially in the brain, is currently out of reach. It would be reasonable to target mitochondrial dysfunction at large, but several trials of mitochondrial treatments for PD have already failed.
Literature
[1] Heger, L. M., Gubinelli, F., Huber, A. J., Cardona-Alberich, A., Rovere, M., Matti, U., … & Burbulla, L. F. (2026). VMAT2 dysfunction impairs vesicular dopamine uptake, driving its oxidation and α-synuclein pathology in DJ-1–linked Parkinson’s neurons. Science Advances, 12(7), eadz5645.
[2] Choong, C. J., & Mochizuki, H. (2022). Neuropathology of α‐synuclein in Parkinson’s disease. Neuropathology, 42(2), 93-103.
[3] Rodriguez-Oroz, M. C., Jahanshahi, M., Krack, P., Litvan, I., Macias, R., Bezard, E., & Obeso, J. A. (2009). Initial clinical manifestations of Parkinson’s disease: features and pathophysiological mechanisms. The Lancet Neurology, 8(12), 1128-1139.
[4] Burbulla, L. F., Song, P., Mazzulli, J. R., Zampese, E., Wong, Y. C., Jeon, S., … & Krainc, D. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science, 357(6357), 1255-1261.