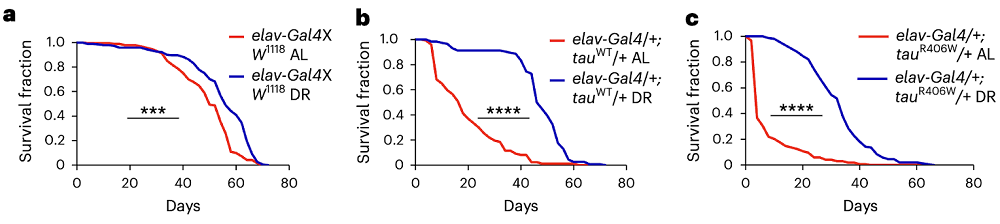
Molecular Similarities Between Cigarette Smoking and Aging
Researchers have analyzed molecular patterns from different tissues obtained from over 700 people and learned that smoking acts as an aging accelerator and involves molecular changes in tissues beyond those directly exposed to cigarette smoke [1].
Millions of preventable deaths
Despite campaigns aimed at the reduction of tobacco smoking, it is still a very common practice and is considered the primary cause of preventable mortality globally, claiming 8 million lives annually [2]. Higher smoking-related mortality stems from an increased risk of respiratory, cardiovascular, metabolic, autoimmune, renal, and infectious diseases along with cancer [3, 4].
Beyond the lungs
Previous studies have addressed the effects of smoking by focusing mainly on airways and whole blood. In this study, the researchers expanded the investigation of the impact of cigarettes on multiple human tissues. They used the Genotype Tissue Expression (GTEx) project, which has data from 46 types of human tissues of 717 individuals, and compared the gene expression in different tissues between smokers and people who never smoked.
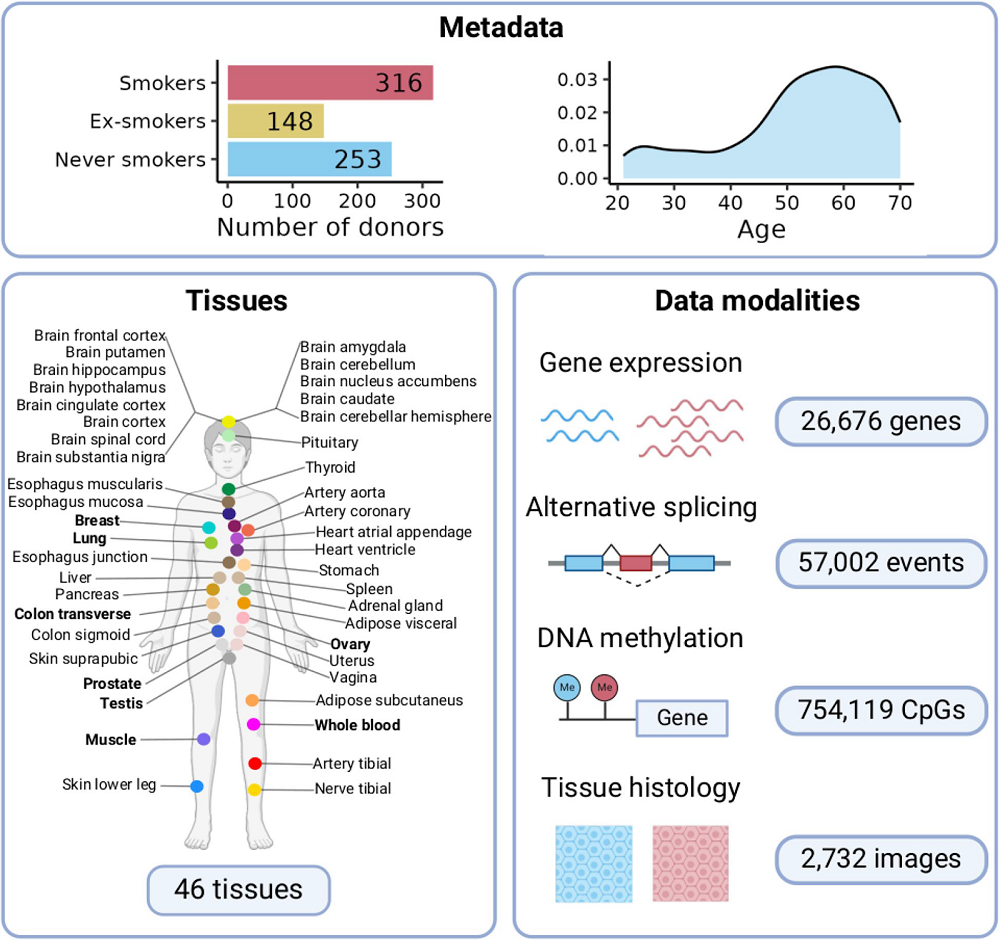
The number of genes differentially expressed between smokers and non–smokers differed depending on the tissue, with most differences occurring in the lungs, pancreas, thyroid, and the cells lining the esophagus. Most of the changes were tissue-specific, and 86% of genes showing smoking-related changes in expression were altered in a single tissue, underscoring the importance of tissue-specific studies.
Only a few genes whose expression was upregulated by smoking were common across nine or more different tissues. A subset of those genes was previously reported to be upregulated by direct exposure to polycyclic aromatic hydrocarbons (PAHs) [5], chemicals formed during tobacco smoking. This connection suggests that toxic compounds from tobacco smoking also reach the tissues not directly exposed to the smoke. Another subset of genes altered by tobacco smoking in several tissues is linked to immune system functions and inflammation.
Along with epigenetics, gene expression can be affected by splicing changes. Genes consist of coding regions (exons) interspersed with non-coding regions (introns). When the DNA of a given gene is turned into RNA, coding regions are spliced together. However, this splicing doesn’t always happen in the same order, and sometimes, not all exons are spliced. This can affect the resulting proteins.
The researchers of this study observed alternative splicing events in 17 tissues from tobacco smokers, with the lung, thyroid, and heart being the most affected. About half of the alternative splicing led to the inclusion or exclusion of an exon, leading to changes in the protein. The other half of alternative splicing events led to the loss of properly coded functional proteins.
Further analysis was focused on the four tissues that showed the most smoking-related changes in gene expression: lung, thyroid, pancreas, and esophagus mucosa. An analysis of images from those tissues suggested structural changes, including at the cellular level. For example, in thyroid tissue, the researchers observed bigger colloid-containing follicles, the storage units of inactive thyroid hormones, which is consistent with the previously reported association between smoking and irregular growth of the thyroid gland [6]. Researchers suggest that the thiocyanate present in cigarette smoke might play a role here, as it inhibits iodine uptake by the thyroid gland, leading to problems with the production of thyroid hormones; however, this was not directly tested.
Inflammatory changes
Previous research observed similarities between gene expression changes in smoking and aging in the respiratory tract [7]. These researchers extended the analysis to different tissues. Eight tissues showed that the overlap between aging and smoking-related differentially expressed genes is higher than would be expected by chance. The changes in gene expression are in the same direction, with many genes associated with the immune system and inflammation.
Beyond these changes in gene expression, smoking also induced changes in methylation patterns. Comparing methylated sites to gene expression patterns revealed that, for the most part, smoking impacted DNA methylation and gene expression independently. However, there were also some shared patterns between the genes whose expression is smoking-associated and the smoking-related hypomethylation pattern. In both groups, the researchers noted enrichment in immune system-related functioning changes, suggesting immune system activation.
Beyond associations
Most observations described so far in this study were associations, not causal links. To establish causality, the researchers turned to the results of a previous study that identified specific methylation sites that have a causal effect on aging-related phenotypes [8]. Overlapping identified smoking-related methylation patterns with methylation sites that have a causal effect on aging-related phenotypes, and there was a substantial overlap in the lung tissues. Those results suggest a causal effect between cigarette smoking and accelerated tissue aging, which acts through DNA methylation of sites that have a causal impact on aging.
Further analysis of different methylation sites by a few epigenetic clocks suggested that age acceleration in the lung results from perturbations at protective methylation sites, that is, sites that contribute to healthy longevity.
Partial reversibility of smoking
Smokers are always advised to quit to improve their health outcomes; however, does quitting impact gene expression changes and DNA methylation patterns? These researchers used data from smokers and never-smokers and compared it to people who stopped smoking. This analysis suggested partial reversibility among most genes, splicing, and methylation events. However, the researchers observed expression changes to more reversible genes than non-reversible ones, making ex-smokers more similar to people who never smoked in terms of gene expression. In DNA methylation, there were fewer reversible sites than non-reversible ones, making ex-smokers more similar to smokers.
Analyzing the effects on gene expression and DNA methylation that are shared between smoking and aging, the researchers noted that in people who quit smoking, non-reversible DNA methylation sites in the lung were enriched in DNA methylation sites that are associated with aging signatures, but this wasn’t the case for reversible and partially reversible sites suggesting “that the smoking effects that affect DNA methylation in common to aging are more persistent in time.” This was not the case for gene expression changes.
Aging accelerator
Taken together, the results of this study support the hypothesis that smoking leads to accelerated aging, with dysregulation of the immune system and inflammation having a strong impact on both processes. While quitting can help reverse some of the smoking-related changes, there are molecular signatures that might persist for a long time.
Literature
[1] Ramirez, J. M., Ribeiro, R., Soldatkina, O., Moraes, A., García-Pérez, R., Oliveros, W., Ferreira, P. G., & Melé, M. (2025). The molecular impact of cigarette smoking resembles aging across tissues. Genome medicine, 17(1), 66.
[2] GBD 2019 Tobacco Collaborators (2021). Spatial, temporal, and demographic patterns in prevalence of smoking tobacco use and attributable disease burden in 204 countries and territories, 1990-2019: a systematic analysis from the Global Burden of Disease Study 2019. Lancet (London, England), 397(10292), 2337–2360.
[3] Thun, M. J., Carter, B. D., Feskanich, D., Freedman, N. D., Prentice, R., Lopez, A. D., Hartge, P., & Gapstur, S. M. (2013). 50-year trends in smoking-related mortality in the United States. The New England journal of medicine, 368(4), 351–364.
[4] Carter, B. D., Abnet, C. C., Feskanich, D., Freedman, N. D., Hartge, P., Lewis, C. E., Ockene, J. K., Prentice, R. L., Speizer, F. E., Thun, M. J., & Jacobs, E. J. (2015). Smoking and mortality–beyond established causes. The New England journal of medicine, 372(7), 631–640.
[5] Stading, R., Gastelum, G., Chu, C., Jiang, W., & Moorthy, B. (2021). Molecular mechanisms of pulmonary carcinogenesis by polycyclic aromatic hydrocarbons (PAHs): Implications for human lung cancer. Seminars in cancer biology, 76, 3–16.
[6] Wiersinga W. M. (2013). Smoking and thyroid. Clinical endocrinology, 79(2), 145–151.
[7] Choukrallah, M. A., Hoeng, J., Peitsch, M. C., & Martin, F. (2020). Lung transcriptomic clock predicts premature aging in cigarette smoke-exposed mice. BMC genomics, 21(1), 291.
[8] Horvath S. (2013). DNA methylation age of human tissues and cell types. Genome biology, 14(10), R115.