Short Summary:
Blood stem cells (HSCs) become dysfunctional with age, impairing our immune systems and our physical function. Two new studies propose opposite solutions to this problem: destroying aged, dysfunctional HSCs or delivering more of the “youthful” minority of HSCs that are found even in aged organisms. Either or both of these strategies could be viable rejuvenation therapies for humans
Imagine waking up after your first comprehensive panel of rejuvenation biotechnologies. At the forefront of your mind would be the treatments’ effects on how you interface with the world: your ability to move and lift your body without pain, see and hear and think clearly, and open jam jars. Next, you’d probably find yourself considering the repairs that, while hidden from your immediate perception, would greatly impact your long-term health: the return of organs like your brain, heart, liver, and kidneys to full, youthful function.
But if you follow longevity science, you’ve likely encountered some remarkable findings about the pro- and anti-aging effects of the blood that connects them all together. This month’s blog post is about a striking pair of studies about a new riff on the theme of “young” and “old blood,” and two opposite (but not mutually exclusive) strategies for solving the problem. Either we can destroy dysfunctional cells that accumulate with age (ApoptoSENS), or we can transplant more of the “youngish” cells that survive into old age to reinforce their youthful function (RepleniSENS).
This teaser may remind you of one of the longest-studied longevity therapies: “heterochronic parabiosis,” in which scientists surgically join a young and an old animal together such that they share one another’s circulatory system. For a short window of time, multiple functions in the aged animal are temporarily “rejuvenated,” even as the young mouse becomes functionally “older” as its blood is partially replaced by that of its older pairmate.
In recent decades, renewed interest in these findings sent many scientists and startups chasing after hypothetical “youth factors” presumed to be present in the blood of young animals that might be turned into a longevity therapy. But surprisingly, an initial a 2015 study funded by the Lifespan Research Institute (LRI — then SENS Research Foundation) yielded compelling evidence that the real key to the parabiosis phenomenon is not any “pro-youth” factors present in the blood of young organisms, but “pro-aging” factors in the old animal’s blood.
A series of followup studies by parabiosis pioneers Mike and Irina Conboy have greatly strengthened this reading of the evidence. Most remarkably, diluting an old animal’s plasma with mere saline solution and a biologically inert “filler” protein is enough to achieve rejuvenation equivalent to — and in some ways superior to — the classic stitch-and-share method of parabiosis. A preliminary human clinical trial in aging humans and another in people suffering with neurodegenerative aging of the Alzheimer’s type support these findings.
A key element of the plasma dilution studies, as well as independent work on young plasma injections, is that the treated animals retain their original blood cells: the procedure only dilutes signaling molecules and other factors dissolved in the animals’ blood. That’s the key difference in these new studies: two groups of scientists, coming at the problem from two different angles, report the harmful effects of old stem cells whose progeny course through the blood, and the hidden “youth” in a subset of them.
Aging Organs One by One
Before we get to the latest findings, let’s review a remarkable study from a couple of years ago that illustrates the importance of blood cells to aging. Laura Niedernhofer at the University of Minnesota has reported out a series of experiments in which she engineers mice with mutations in their cells’ DNA repair machinery, but only in specific organs. Cells in the affected organs in such mice develop high levels of unrepaired DNA damage, and in some (but not all) organs, a lot of these cells pull the “emergency brake” by going senescent. This experimental design allows Dr. Niedernhofer to look at the effects of the resulting cellular damage (most prominently, a high burden of senescent cells) in a single organ, in a mouse that is otherwise young and healthy.
In one of these experiments, Dr. Niedernhofer put this mutation into haematopoietic stem cells (HSCs) of mice. HSCs are the specialized stem cells responsible for producing all the different cell types in the blood, including red blood cells and immune cells. These animals seemed healthy enough until they reached early adulthood — but then their immune systems started to crash. First they started to lose T-cells, and by midlife their B-cell numbers also begin to plummet.
All of these results were fairly predictable from the fact that the stem cells for the animals’ blood cells couldn’t repair their DNA properly. With all that DNA damage, multiple kinds of immune cells were already senescent by the time the animals became young adults — and it only got worse from there, especially for their macrophages.
But the really noteworthy results came when the researchers looked at the other organs in these mice. Remember, only the blood cell precursors in these mice were engineered to have defective DNA repair: all their other organs had normal DNA maintenance. Yet when Niederhofer and colleagues looked at the other tissues in these mice, they found that the animals already had very high levels of senescent cells by the time they were middle-aged — especially the liver, pancreas, and large blood vessels, with mixed signals in the gut and lungs. They also had elevated markers of liver damage, and levels of the cocktail of key blood-borne senescent cell secreted factors (SASP) that in some cases rivaled those of geriatric mice.
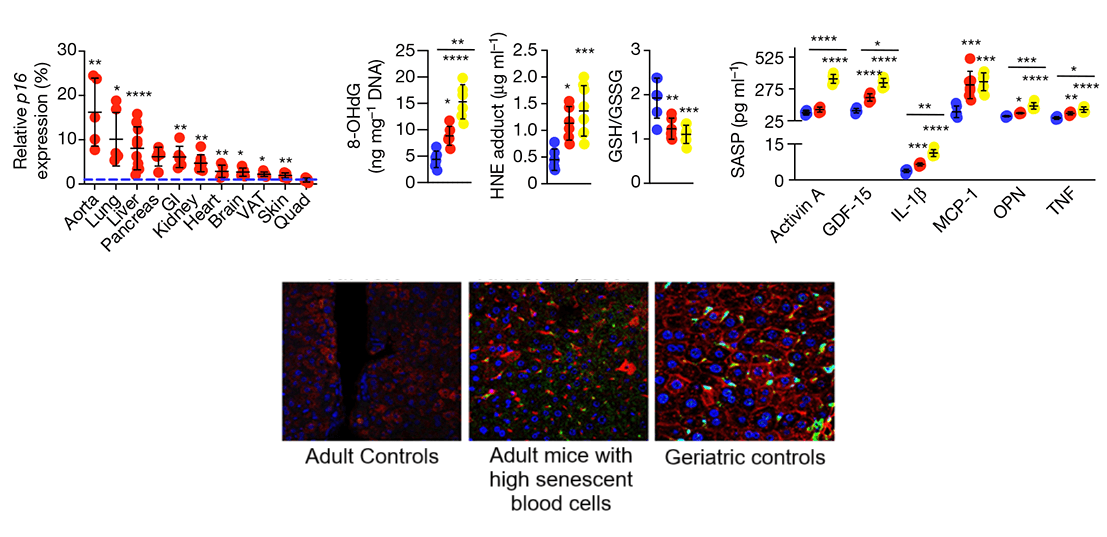
A lot of this harm likely stems from one of the more recently-discovered and insidious effects of senescent cells: secondary senescence. As we’ve discussed before, the SASP proteins secreted by senescent cells do more than break down local tissues’ barriers against cancer and make the immune system go haywire with inappropriate inflammatory molecules: they also drive non-senescent cells into senescence. In this case, the fact that the legions of senescent cells were circulating in the blood rather than in a fixed location in some solid organ doubtless gave the senescent blood cells an exceptional sweep across the body, although even solid organs from old animals can spread senescence to remote tissues.
Still, the investigators wanted to make sure that the high senescence burden in these animals’ solid organs was the downstream result of their damage-prone blood cells, and not (for instance) due to some defect in the engineered gene that caused it to unintentionally activate in the mice’s other organs. So the investigators drew the mutation-bearing animals’ blood, destroyed all the red blood cells (which don’t become senescent*), and transfused this blood into young, normal (wild-type) mice.
In this experiment, then, the only cells with the repair mutation in the transfused animals were blood cells from the mutation-bearing mice, ruling out any unexpected effect of the transgene in non-blood cells. Moreover, the mutation-bearing blood cells could only begin harming the normal mice once they were transfused, whereas the mutation-bearing mice’s own blood cells were potentially already harming the rest of their tissues, albeit that there was no obvious evidence of ill effects until the mice were young adults. And as an additional reality check, the researchers infused another group of normal young adult mice with the blood of quite old but mutation-free mice, to see if the effects of the “accelerated aging” mice’s blood were really reflective of the impact of “normal” blood cell aging.
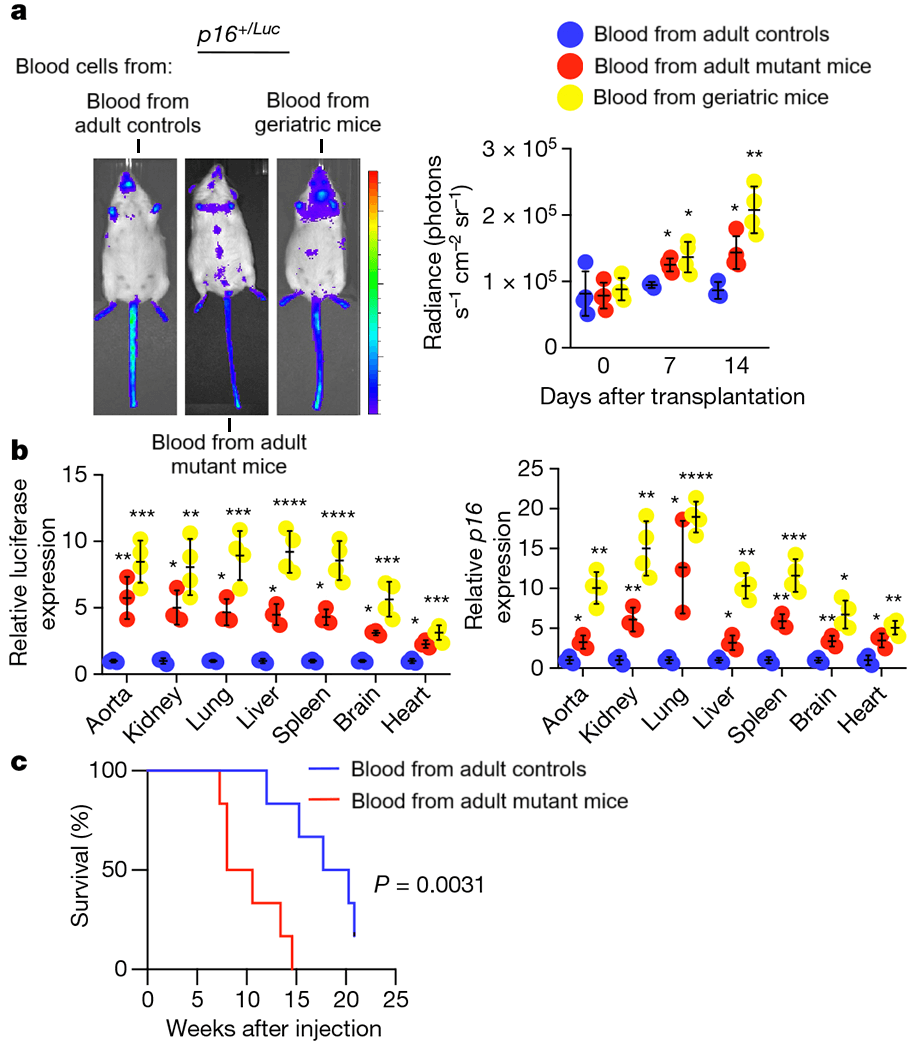
So we know that there are harmful factors borne in the aging blood, including both soluble signaling factors (as shown by the plasma exchange studies) and aged (likely senescent) immune cells (in Dr. Niedernhofer’s mice). But we also know from looking at the organs of aged mice and humans that only a small fraction of the cells in a given tissue go senescent with age. And additionally, we know that a lot of things go wrong in aging immune cells other than senescence, leaving us vulnerable to age-related disease and debility in ways that are not obviously connected to the failure to fight off microbial invaders. Might there be other bad actors in need of purging — or good actors in failing numbers and in need of replacement?
The answer to both questions seem to be “yes,” according to two recent studies in aging mice.
Separating the Sheep from the Goats
We mentioned earlier that HSCs produce our immune cells. The two new studies on HSC aging are focused on the differential effects of aging on the two broad categories of immune cells: the myeloid cells and the lymphoid cells. Lymphoid cells (like T and B cells) are mostly highly specialized assassins, responsible for adapting to new threats (hence, the “adaptive immune system”). Through molecular intelligence reports from other immune cells, they learn to identify a specific bad actor that has entered the body and then seek out and destroy it. When the battle is over, a small cadre of veterans of this first encounter with the enemy are held in reserve, able to immediately recognize their old enemy the moment it rears its head. The lymphoid cells are thus responsible for the long-term immunity we get from vaccines or post-infective immunity.
By contrast to this hyperspecialization, myeloid cells primarily act like sentries on patrol, looking for anyone who looks to be somehow suspicious. They can mount a response early, before the adaptive immune system even has a chance to figure out what it’s up against, but their attacks are accordingly more generic, and they don’t retain a memory of any specific threat for the future. This nonspecific activity makes up the “innate” immune system. The scouting activity of some myeloid cells also relays information about enemy combatants back to the lymphoid system so that they can get into the fight.
Youthful immune function requires the concerted action of both myeloid and lymphoid immune cell types. But with age, HSCs become like loaded dice, with rising numbers of HSCs biased toward forming myeloid cells, leaving fewer HSCs to form lymphoid cells. At a minimum, this “myeloid skew” puts us at greater risk of problems driven by overly-assertive myeloid cells (such as atherosclerotic cardiovascular disease (ASCVD), chronic inflammation, and some blood cancers), while simultaneously leaving us more vulnerable to infections (like COVID) and other threats for which a substantial supply of lymphoid cells is an essential defense. (The deficit is most profound for the not-yet-specialized “naïve” T-cells).
Fifteen years ago, a group of researchers that included Irv Weissman (one of the original discoverers of the myeloid skew with age) went spelunking among the HSCs of aging mice and discovered that myeloid-tilted HSCs have a marker on their surfaces called CD150. By selecting HSCs with low levels of CD150 on their surfaces and transplanting these cells into aged mice, they were able to rebalance the age-related myeloid skew in these mice, even if the “low-CD150” cells originally came from old mice. In other words, the skew was largely confined to a specific subset of cells, not some universal property of all HSCs in aging organisms.
This finding was a good proof-of-principle, but not an easily workable therapy for aging people: you would have to harvest a person’s bone marrow, purify out the relatively small number of low-CD150 HSCs, grow out large numbers of them in a lab, and deliver them back to their host via a bone marrow transplant.
Just recently, researchers Jason Ross and Lara Myers at Rocky Mountain Laboratories teamed up with Weissman to ask if there was a simpler solution: instead of wiping the slate clean and replacing all a person’s HSCs with the functionally “younger” subset, could one correct age-related myeloid skew by simply destroying large numbers of myeloid-biased HSCs?
Because myeloid-biased HSCs (my-HSCs) have high levels of CD150 on their surfaces, the researchers injected adult or middle-aged mice with antibodies that would bind to CD150, thereby tagging myeloid-skewed HSCs for destruction — either by immune cells that would recognize the antibody and destroy the associated cell, or by engaging other immune mechanisms. To boost the effect, they simultaneously treated the mice with an antibody that would block a cell-surface protein called CD47. This protein sends a “don’t eat me” signal to ward off immune cells that would otherwise swallow cells with antibodies on them: that way, if any my-HSCs were flagged with CD47, such immune cells would ignore their protestations and gobble them up.
This protocol (as well as several other antibody combinations the researchers tested) quickly knocked down the number of my-HSCs in young adult mice. Even in these relatively young animals, their stem cell balance became more youthful-looking after treatment. And just one week after treatment, the mice’s rebalanced HSC population produced a better balance of committed progenitor cells —the cells one step further along the developmental pathway from HSCs toward mature immune cells, which are fully “committed” to become lymphatic or myeloid cells rather than being merely “biased” one way or the other. More cells “committed” to becoming lymphoid cells or retained the potential to go either way, and fewer committed to becoming myeloid cells. Accordingly, the ratio between committed lymphoid to myeloid progenitors increased by up to four times that of untreated mice.
To verify that the effects of the depletion antibody were due to the rebalancing of the HSC population (and not because they were unwittingly helping with some other problem in the animals’ aging bodies), the investigators first used chemotherapy to wipe out the HSCs from very young mice, and then transplanted a tiny number of HSCs from either antibody-treated or -untreated middle-aged mice. Sure enough, HSCs from the antibody-treated mice repopulated the young transplant recipients’ stem cell compartments with a much more balanced HSC pattern than when mice got HSCs from untreated mice. And animals that received “purged” HSCs produced a youthful balance of progenitor cell types, while animals that got HSCs from untreated animals produced an “old”-looking excess of myeloid progenitor cells and not enough lymphoid progenitors.
But none of these studies had lasted more than a week. You might worry that the aged animals’ HSC balance might look youthful in the immediate aftermath of a purge, only to quickly snap back into its old, dysfunctional pattern as their remaining my-HSCs multiplied furiously to restore the old imbalance, leaving the animals no better off than they were before treatment.
Also, all these experiments were done in relatively young mice, so you might wonder if depleting my-HSCs might not work in truly old mice. For instance, what if some form of cellular or molecular aging damage responsible for the myeloid skew in an aged organism’s immune cells was independent of the my-HSCs themselves, such that a rebalanced population of HSCs would still lead to a defective repopulation when performed in an aged body? As a more specific example: what if aging processes damage the bone marrow in such a way that it can no longer nurture lymphoid-biased or balanced HSCs, no matter how much you purge the myeloid-skewed cells?
To answer these questions and test the clearance approach under conditions that would mimic its use as a rejuvenation biotechnology for humans, the Rocky Mountain Labs researchers used their antibodies to deplete my-HSCs from either young adult mice or mice that were broadly “old,” the latter group including mice ranging from late middle mouse-age to geriatric.† And this time, they tested the longer-term effects of HSC depletion one and four months after treatment (remembering that two months is a fraction of a mouse’s lifespan similar to a decade of human aging).
One week after treatment, the number of my-HSCs in the old mice had plummeted, but the number of lymphoid-committed progenitors had not yet begun to improve. Over the next four months, the my-HSCs started to slowly creep back up again — but in the meantime, the lymphoid progenitors multiplied even more quickly, leading to a net rebalancing of the population.
Next, the researchers looked for the effects of the depletion on the animals’ production of “naïve” T- and B-cells — that is, new T- and B-cells that have recently emerged from their maturation centers in the body and haven’t yet encountered a pathogen. We lose naïve lymphocytes in our bodies with age, while we accumulate “memory” cells, which are the descendants of T- and B-cells that previously encountered a pathogen and are held in reserve in case the same invader comes back for another round.
At the two-month mark (which is how long it takes for HSCs to progressively mature into new B- and T-cells), the treated animals had modestly more naïve T-cells than did untreated animals, and enjoyed a wider normalization of other T-cell subpopulations. This was an important finding: the age-related loss of naïve T-cells is likely one key reason why older people are so vulnerable to new pathogens, as we saw with the ravaging of our elderly by COVID and continue to see with new variants of the flu.
And this surge of new naïve T-cells occurred despite the lack of any effect on the mass of the thymus — the organ responsible for maturing T-cells. The thymus normally shrivels with age, losing its functional tissue in favor of fat cells, and scientists generally point to this shrinkage as the main thing responsible for the age-related loss of native T-cells in mice (although this no longer seems to be as important a factor in humans as it is in mice). The fact that naïve T-cells were boosted by my-HSC depletion suggests that the age-related myeloid skewing of the HSCs themselves is also an important driver, independent of what happens to the thymus, contributing to our inability to mount effective defenses against new immune threats as we age.
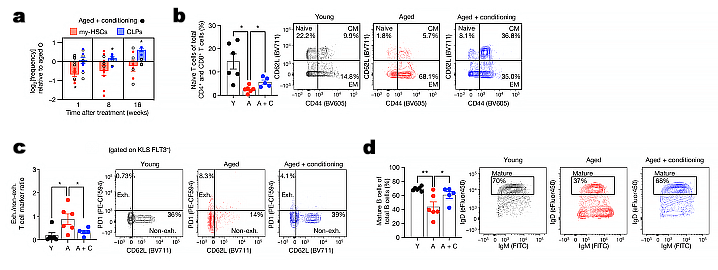
The treated animals also enjoyed a reduction in two key dysfunctional immune cell populations that accumulate with age: “exhausted” T-cells and Age-Associated B-Cells (ABCs). Similarly, the treated mice enjoyed a near-complete restoration of their B-cells to youthful levels.
Researchers who study immune aging generally think that myeloid immune cells like macrophages are responsible for much of the rise in inflammation with age: this was the origin of the whole idea of “inflammaging,” long before scientists discovered that senescent cells produce inflammatory signaling molecules as part of the SASP and implicated it in the process. Sure enough, mowing down the excess my-HSCs in aged mice reduced their levels of multiple markers of inflammaging; in fact, the two most profoundly elevated markers fell back to normal, youthful levels.
Rejuvenated Immune Defense
The fact that the treated mice had a more youthful pattern of immune cell types and lower inflammation is all well and good — but what we really want to know is whether destroying excess my-HSCs actually improves immune function. To approach this critical question, the Rocky Mountain Labs scientists first treated aged mice with either a dummy injection or antibodies to cull my-HCS p aged mice’s immune function. They then vaccinated both of these aged groups and a young control group against the FV mouse virus, for which an effective immune response requires both B- and T-cell responses. Unvaccinated old and young mice served as additional controls. They then gave all the mice an additional six weeks to “staff up” their immune reserves before infecting groups of them with real FV mouse virus.
Even in young animals, unvaccinated mice’s spleens swelled with infected immune cells, with a median of 7,000 such cells per spleen — but vaccination almost eliminated this upswell. By contrast, some four million of the unvaccinated old mice’s immune cells became infected, and while vaccination helped, it only brought the vaccinated mice’s infected cell counts down to the level of unvaccinated young mice, and only one out of eight mice ultimately got the infection under control. But when old mice had their my-HSCs purged, the vaccine was able to cut the number of infected cells by another 50% or so, leaving them halfway between unvaccinated and vaccinated young mice, and four out of nine mice fully cleared the bug.
In Through the Out Door
At about the same time that the Stanford-Rocky Mountain collaboration was discovering the benefits of depleting dysfunctional my-HSCs, a separate group from Harvard was working on the same problem from the opposite direction.
Dr. Yuting Wang and colleagues were following up on the many studies that had revealed benefits of transplanting young HSCs into old animals beyond those you would expect from just increasing their ability to make new blood cells. The team first dug further into the intervention’s effects on the immune repertoire, showing that young-to-old HSC transplantation restores a more youthful immune cell profile to aged mice. Although the two groups were working in ignorance of one another, this result was strikingly similar to what the RML scientists had achieved with my-HSC depletion: the excess of myeloid cells declined, the number of naïve T-cells increased, and several other immune cell populations changed in a youthful direction parallel to the depletion study.
They then showed that the young HSCs conferred some surprising benefits beyond the blood, boosting the old animals’ strength, coordination, general activity, and some but not all cognitive functions.
What might have gone so wrong with the old animals’ HSCs? To probe this question, Wang and colleagues compared the gene expression in the old animals’ HSCs to those of young mice. They found that while the young mice’s HSCs were all busy expressing the same well-defined set of genes, the old mice’s HSCs contained three distinct subpopulations, each with its own gene expression pattern. This either suggests a general loss of some of the old animals’ HSCs’ intrinsic ability to properly carry out their role, and/or that one or the other of the aged mouse subpopulations was adapting (for better or for worse) to some kind of damage outside of themselves in the old organism. And yet, while two of these subpopulations were quite different from the HSCs of young animals and also from each other, the third subpopulation of HSCs in the old animals retained nearly the same gene expression patterns as did the HSCs of young mice.
To drill down into what makes these populations different, the Harvard scientists sought a way to conveniently separate the “young” HSCs in the old animals from the two “old” subpopulations. They compared the three populations’ expression of genes that code for proteins present themselves on the surface of cells, as an antibody would be able to bind such cells, which would allow Dr. Wang and colleagues to separate the young-looking HSCs from the ones that were acting their chronological age. And just like the RML scientists, they found that the two old HSC subpopulations had much higher expression of CD150 on their surfaces than did the more youthful-looking HSC subpopulation. (They found other cell-surface proteins that could also have done the job in principle, but CD150 has the advantage that antibodies against it are readily available).
Wang and colleagues then used an antibody that binds CD150 to pluck functionally older HSCs out of the overall HSC population of old mice. As expected, high-CD150 HSCs from old mice looked old: as compared to HSCs from young mice, they exhibited epigenetic changes, more genetic damage, more signs of stress from intracellular aggregates, and less ability to replicate. Interestingly, however, low-CD150 HSCs from the same mice looked much more like the HSCs from young animals.
They then tested the different HSC subpopulations’ classical stem cell activity: the ability to serially repopulate the bone marrow of mice when used for transplant. HSCs from young animals seem to have an almost limitless ability to do this. You can take a young mouse’s HSCs, transplant them into another mouse whose own HSCs have been destroyed by chemotherapy or radiation, and watch the HSCs rebuild the bone marrow of the transplantee and start producing all the appropriate blood cells again. What’s more surprising: you can then take the HSCs from that second mouse and transplant them into a third mouse and watch the whole cycle repeat itself … and then again into a fourth mouse. On and on.
But old mouse HSCs largely lose this remarkable ability. So Wang and colleagues sought to test the relative capacities of the high- and low-CD150 HSC subpopulations of old mice to carry out this feat. The “young” (low-CD150) HSCs from old mice weren’t quite as functional as the HSCs of young animals — after two rounds of serial transplantation from one animal to the next, they had about three quarters of the ability of a truly young animal’s HSCs to repopulate the last animal’s bone marrow — but they beat the stuffing out of the same animal’s high-CD150 HSCs, which were only 5.9% as effective. Conversely, the few high-CD150 HSCs that Wang and colleagues could scrounge up in the bone marrow of young animals were modestly weaker than the dominant low-CD150 HSCs from these animals, again showing that CD150 expression doesn’t fully correspond to an HSC’s youthful function.
How Old Do You Think I Am?
All of this evidence that old animals retain a small reserve of HSCs that are still youthful amid a crowd of dysfunctional cells led the Harvard team back to where they had started. If transplanting young HSCs into old animals can rejuvenate the old ones in several ways, could young-looking low-CD150 HSCs from old animals do the same rejuvenating job?
To answer this question, Dr. Wang and colleagues gave middle-aged mice HSC transplants from old animals (similar to human 70-year-olds), using either “young-looking” HSCs selected for low CD150 expression, “old-looking” HSCs with high CD150, or the old animals’ all-comers HSC population. Five months after transplanting these cells into middle-aged mice, the Harvard researchers looked at the animals’ blood and tested their physical and cognitive function. (Bear in mind that five months of a mouse’s life is the equivalent of about fifteen years of human aging). Their expectation — and likely their hope — was that the low-CD150 HSCs would deliver the same rejuvenating effects that they had previously seen when they transplanted older animals with young animals’ HSCs.
But while there were definitely differences among the effects of the various populations of HSCs, there turned out to be nothing special about the low-CD150 HSCs. On most measures, there was no real difference between the mice transplanted with low-CD150 HSCs and those transplanted with the whole, unselected HSC population of old mice. Rather, the only consistent difference was that the mice given HSCs with the “old” profile (high CD150 levels) fared worse than one or the other or (usually) both of the alternative transplant regimens. This suggests that while expressing high levels of CD150 on a cell’s surface is a marker of age-related dysfunction, merely having low levels of this protein on an HSC’s cell surface doesn’t mean that an HSC has escaped the ravages of time, despite the youthful-looking differences in the expression of other genes.‡
But surprisingly, this pattern reversed itself when the researchers looked at the effects of the different HSC transplantations on the mice’s lifespans. Instead of just testing these animals’ blood and physical function five months after transplantation and then sacrificing them, they let the mice live out the rest of their lives. And contrary to what they’d seen with individual functional measures, that single transplantation of low-CD150 HSCs from old animals into middle-aged ones quite substantially increased their lifespan, while there was no difference between animals given unsorted old HSCs and the older-looking high-CD150 subpopulation of HSCs.
This result is likely more in line with what the investigators had originally hypothesized would happen for the tests they ran at the five-month mark, but instead they(?) saw no benefit to the low-CD150 transplantees. Even more surprisingly, the mice didn’t start dying until many months after they received the single transplant, and the last of the low-CD150-transplanted animals burned down its candle some twenty months later — as long as the entire lifespan of some mice.
(For the record, the Harvard scientists could have made this study much more informative if they had also included a group that received HSCs from young animals, to directly compare their effects with the “young-looking” (low-CD150) HSC subset from old animals, as well as a control group of animals not given any transplant or (better yet) given a sham treatment. Indeed, throughout the paper, one can see places where another comparison group would have strengthened or clarified the results. You can get some sense of what the results might have been for some tests by comparing different experiments in their paper, but you’d ideally want to have all the transplant regimens (young, high-CD150 old, low-CD150 old, all-comers old, and sham-treated controls) conducted in each experiment, which is not what we got in this paper).
When You Come to a Fork in the Road, Take It!
These two new studies take opposite approaches, but point us toward some shared conclusions about the aging and rejuvenation of the HSC compartment. They both tell us that the HSC population of old mice is a mix of relatively “young” and functional cells with other cells that are decidedly not. The more aged population is merely impaired in some functions (Harvard) but is actively harmful in other ways (RML). Either destroying the more aged HSCs or supplementing more youthful ones can not only restore HSCs’ core stem cell function, but also rejuvenate the immune cell population and function of aging mice.
The most impressive outcome of this rebalancing of the immune system is the bounceback of the aging mice’s flagging population of naïve T-cells. The production of these cells slows to a trickle with age — a fact that has traditionally been attributed to the atrophy of the thymus gland (although this no longer seems to be as important a factor in humans as it is in mice). These cells are critical to our ability to take on new microbial threats, so the fact that both of these strategies restore naïve T-cell counts without any effect on the thymus is a very pleasant surprise.
Both strategies also lead to rejuvenating effects beyond the blood in old mice. The young-HSC-transplant strategy demonstrated more such effects than the depletion strategy, although we don’t know whether depletion of the aged HSC population would have the same effects because, in most cases, the RML scientists didn’t test for these outcomes. And both studies showed that scientists or future doctors can use a relatively simple biotechnology (monoclonal antibodies) to either sort out the two populations or to destroy the dysfunctional subset.
Of the two, destroying dysfunctional cells (ApoptoSENS) is more straightforward and seems achievable on a shorter timeframe. Antibodies against CD150 are already a common reagent for laboratory use, though for use in human medicine the manufacturing process would have to be brought up to Chemistry, Manufacturing, and Controls (CMC) standards. Then, it would be a matter of performing animal and human studies to optimize the dose and running clinical trials with one or two antibodies to see if we can reproduce the rejuvenating effects seen in the mouse experiments in aging humans.
And while it’s speculative, purging these cells might also improve clonal haematopoiesis, the tendency of HSCs with a particular mutation to outcompete genetically intact HSCs and become the dominant HSC population with age. Clonal haematopoiesus seems to increase a person’s risk of some blood cancers as well as atherosclerotic cardiovascular disease (ASCVD), so eliminating these cells would be expected to have significant benefits beyond those seen in the rodent studies. If cells with high levels of CD150 (or one of the other cell-surface proteins that strongly mark my-HSCs) are also disproportionately members of the mutation-bearing HSC clone (which seems like a reasonable hypothesis), then clearing such cells would yield a side-benefit that the RML study didn’t investigate.
Transplanting young HSCs is a much more complex and risky endeavor. First, it’s not likely sustainable to put a person through multiple rounds of transplantation with their own low-CD150 HSCs, as the law of diminishing returns will set in with the aging of a person’s HSCs and the impact of the procedure on the cells.§ Look back at the dropoff between the first and second rounds of serial HSC transplants in the Harvard experiments above, and imagine repeating the exercise (say) every five years.
In principle, we could instead give aging people transplants with HSC donated by young adults; the most obvious barrier to that approach is the limited number of chronologically young donors — a demographic barrier that would get worse over time, creating a cell therapy Ponzi scheme. And in addition to the supply question, there are safety concerns with such an approach. While we’ve been doing bone marrow transplants for six decades now and more than 20,000 transplants of HSCs or related cells are performed in the US every year, they’re still expensive and uncomfortable, with very significant near-term risks during the window between when a person’s immune system has been wiped out and when the transplanted HSCs reconstitute the immune system, as well as longer-term risks from drugs that suppress the immune system, which are often required for such transplants.
The worst of these risks could be reduced by using more frequent rounds of relatively mild “mini-transplants” (non-myeloablative transplants), but at the expense of leaving more of a person’s aged HSCs behind, which would necessitate a more frequent schedule of repeat transplantations, with each round exposing a person to risk anew.
However, the risks and inconvenience would decrease substantially once the still-challenging and experimental process of cellular reprogramming becomes part of clinical medicine. By taking a person’s circulating blood cells, walking them back into an early developmental state, and then nudging them forward to become HSC, the sourcing of cells would become logistically if not technically simple, and all concerns related to immune mismatch and most if not all issues with donor age would disappear. (Indeed, one intriguing question raised by the study by Wang and colleagues is how much of the substantial rejuvenating effects of in vivo partial reprogramming in its latest iterations is due to HSC reprogramming specifically).
HSC transplants with newly-“young” reprogrammed HSC would also enable us to piggyback on the process by engineering the cells to produce additional rejuvenation biotechnologies, such as antibodies to remove extracellular aggregates and enzymes to degrade intracellular aggregates. Turning one’s bone marrow into factories for these therapies would allow for continuous low-level delivery instead of larger periodic doses delivered by infusion, which is likely to be both more effective and to reduce any risks (at minimum, from the infusion itself!), as well as the inconvenience of having to take time out of one’s life to come in to get topped up on such therapies. And over a longer time horizon, HSC replacement with therapeutic modification is a key part of the ambitious WILT strategy for near-exhaustive cancer prevention.
And while destroying dysfunctional HSCs seems like the simpler and nearer-term strategy to implement, we don’t ultimately need to choose: the two approaches are not mutually exclusive, and weeding out dysfunctional cells while also receiving “mini-transplants” of reprogrammed HSCs would improve the latter’s effectiveness and accelerate the turnover of the overall HSC population toward therapeutically-enhanced or WILTed HSCs without having to always resort to riskier full-on bone marrow transplants.
Ah, from the pit of such a garden
I would draw new strength. I would
Lay by the soiled garment of the day.
I would become new, young; mine eyes
Darkly proclaiming their emptiness;
And I, a little child, would stand
Within the garden’s pit,
Letting the words of God’s wisdom
Whisper from the tips of the flower’s tongues,
And I, listening, would no wiser be;
But new dreams of stuff most tenuous would
Have flowed, like a phantom pageantry into me;
And I, looking with my night-dark eyes
Upon the sun, lingering in the deep sky,
Would speak no word of reproaching for
The speeding time; for I, youthed, yea, youthed,
Would know the wisdom of waiting! And tomorrow?
Oh tomorrow! Why, tomorrow is eons hence!
And yesterday? Oh yesterday!
I lost her completely. But today is here
In the garden, and the pit of its sweetness-From The Garden of Youth by Patience Worth
*
Red blood cells become defective at the end of their life, and for historical reasons this happens to be called “senescence,” but it’s a totally separate phenomenon from the cellular senescence involving replicative arrest, DNA damage signaling, and the SASP that we discuss in other cell types..
†
The investigators made a poor decision on this one. The age range is too broad to use when the whole point of the experiment is to look at the effect of age, especially since we don’t know whether the treated or the untreated mice might have included more of the older mice from the outset..
‡
On the other hand, it could be that all the beneficial effects of whole HSC transplant were the result of work done by the low-CD150 subset within it, and that the other HSCs in the mixed population are just along for the ride. But that doesn’t seem likely, because it would mean that a small subset of total cells in the mixed population was just as effective as a fully-purified high-CD150 population.
§
Because the body contains large numbers of HSCs and they need to frequently divide to produce new precursors for blood cells, the situation is quite different from (say) transplanting the neurons that are lost in Parkinson’s disease, which are few in number, don’t divide, and last for many decades.