Towards a Telomerase Gene Therapy for Pulmonary Fibrosis
Back in January 2018, researchers at CNIO reversed lung fibrosis in mice using a single gene therapy. Today, we want to spotlight a new study that builds on the positive results of the 2018 research, taking us another step closer to telomerase gene therapy in humans to reverse pulmonary fibrosis.
The road to a gene therapy for pulmonary fibrosis
Idiopathic pulmonary fibrosis is an age-related disease that currently has no cure and very limited treatment options. Pulmonary fibrosis is a disease of the lung tissue that causes it to become stiff and scarred, leading to a progressive shortness of breath.
This disease seems to develop due to a combination of genetic and environmental factors. While exposure to toxic substances can play a role in its development, the disease typically appears during advanced age or due to an underlying genetic condition, and it is linked to telomere dysfunction.
Telomeres are located at the ends of our chromosomes, which store our genetic information. A telomere contains a specific DNA sequence that is repeated thousands of times and serves as a protective cap, and it is sometimes likened to the aiglet on the end of a shoelace, preventing it from unraveling.
This sequence has two functions: it protects the coding regions of the chromosomes and prevents them from being damaged, and it acts as a clock that controls the number of replications that a cell can make. The telomeres shorten with each cell division, and this erosion is thought to be one of the reasons we age.
Telomerase gene therapy activates the production of the telomerase enzyme, which repairs the telomeres located at the ends of our chromosomes, restoring the sequence and building the protective cap back up. The telomerase therapy used by the Spanish National Cancer Research Centre (CNIO) is based on activating telomerase expression transiently. To achieve this, a virus is used as a telomerase gene carrier and is injected intravenously into the mice.
Telomere dysfunction leads to pulmonary fibrosis
Previous studies by the CNIO team had shown that there are genetic factors linked to the dysfunction of telomeres. In 2015, the team developed a superior mouse model of pulmonary fibrosis that lacked the telomerase gene, which prevented those mice’s cells from expressing telomerase.
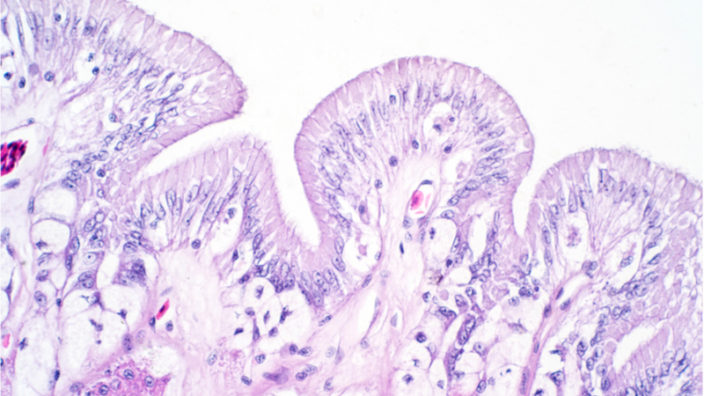
This mouse model works because alveolar type II cells, which play a role in lung tissue regeneration, experience telomere dysfunction and die. This results in the mice developing pulmonary fibrosis as the lung tissue is unable to regenerate and repair itself periodically, resulting in the formation of fibrotic scar tissue.
In 2018, the CNIO team published a study using their superior mouse model of fibrosis and demonstrated that telomerase gene therapy was able to reverse that fibrosis [1].
A new study published by Dr. Maria Blasco and her research team at the Telomeres and Telomerase Group at the CNIO, in collaboration with researchers from the Complutense University of Madrid and the Autonomous University of Barcelona, shows that the same telomerase therapy can successfully treat age-related fibrosis in old mice.
This is an important development because it shows that not only does telomerase therapy work in mouse models of fibrosis, it also works in mice that have not been genetically modified in any way and whose fibrosis is simply the result of normal aging.
These data also put telomere attrition in the spotlight for why the disease develops and show that the primary risk factor for that attrition is aging. Telomeres slowly erode after many cell divisions until they become so short that they cannot protect the chromosomes; at this point, the cell triggers an alarm and ceases to divide as a safety feature. However, once enough cells reach this state, the tissue can no longer regenerate.
This new study shows that, like the mouse model’s engineered cells, age-related telomere attrition naturally occurs in alveolar type II cells, a key player in lung tissue regeneration. The study results strongly suggest that the researchers have discovered the molecular basis of the association between aging and pulmonary fibrosis.
In the new study, the researchers describe the effect that aging has on alveolar type II cells and how that impacts their ability to function and facilitate tissue repair and regeneration. As well as regenerating the lung tissue, alveolar type II cells also secrete a lipid-protein complex called pulmonary surfactant, which lubricates the lung tissue, helps it stay elastic as it expands and contracts during breathing, and reduces the energy required to breathe.
This means that if the alveolar type II cells are unable to regenerate, then the pulmonary surfactant will not be produced, which causes the lung tissue to become stiff and fibrotic. The researchers show that there is a direct correlation between telomere function in alveolar type II cells, pulmonary surfactant secretion, and the development of fibrosis in animals.
Short/dysfunctional telomeres are at the origin of idiopathic pulmonary fibrosis (IPF) in patients mutant for telomere maintenance genes. However, it remains unknown whether physiological aging leads to short telomeres in the lung, thus leading to IPF with aging. Here, we find that physiological aging in wild-type mice leads to telomere shortening and a reduced proliferative potential of alveolar type II cells and club cells, increased cellular senescence and DNA damage, increased fibroblast activation and collagen deposits, and impaired lung biophysics, suggestive of a fibrosis-like pathology. Treatment of both wild-type and telomerase-deficient mice with telomerase gene therapy prevented the onset of lung profibrotic pathologies. These findings suggest that short telomeres associated with physiological aging are at the origin of IPF and that a potential treatment for IPF based on telomerase activation would be of interest not only for patients with telomerase mutations but also for sporadic cases of IPF associated with physiological aging.
Conclusion
This research holds great potential and may offer a solution to all types of fibrotic diseases. The next steps will be to translate these findings to people, and we are confident that human trials for telomerase gene therapy are not too far away now.
It has taken over a decade of steady research by Dr. Blasco and the CNIO, but we are finally reaching the point where telomerase therapy could soon be ready for prime time. It will be a truly exciting day when we can finally report the launch of the first human trials for telomerase therapy, and let’s hope that day is soon.
Literature
[1] Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. Maria Blasco, Juan Manuel Povedano, Paula Martinez, Rosa Serrano, Agueda Tejera, Gonzalo Gomez, Maria Bobadilla, Juana Maria Flores and Fatima Bosch (eLife 2018). DOI: 10.7554/eLife.31299