New Study Calculates Lifespan Gains From Five Popular Diets
Scientists have pitted five diets against each other to see which one is associated with more years of life gained [1].
The clash of the diets
Unhealthy eating is recognized as a globally leading cause of death [2]. Surprisingly, few studies have actually evaluated the gains in life expectancy associated with adherence to a healthy diet. In a new study published in Science Advances, an international group of scientists pitted five leading dietary practices against each other using data from UK Biobank, a huge repository of health-related information on hundreds of thousands of British citizens.
The sample was comprised of 103,649 participants (mean age 58.3 years, 56.4% female) who had completed two or more web-based 24-hour dietary assessments and were free of cardiovascular disease (CVD) and cancer at baseline. Each participant was scored on five dietary pattern indices based on what they reported eating: Alternate Healthy Eating Index (AHEI-2010), Alternate Mediterranean Diet (AMED), healthful Plant-based Diet Index (hPDI), Dietary Approaches to Stop Hypertension (DASH), and Diabetes Risk Reduction Diet (DRRD).
Each score was divided into quintiles. The five scores were moderately-to-highly intercorrelated, meaning that the dietary patterns they capture are often overlapping (which is expected), but not identical. The model was adjusted for race, education, socioeconomic deprivation, smoking status, physical activity, BMI, total energy intake, baseline dyslipidemia, hypertension, diabetes, and, for hPDI, DASH, and DRRD, alcohol consumption.
The researchers also wanted to see how dietary patterns interact with known longevity gene variants. They calculated a longevity polygenic risk score (PRS) from 19 single nucleotide polymorphisms (SNPs) which were significantly associated with longevity in a genome-wide association study (GWAS). This model was additionally adjusted for ten genetic principal components.
The winners and the losers
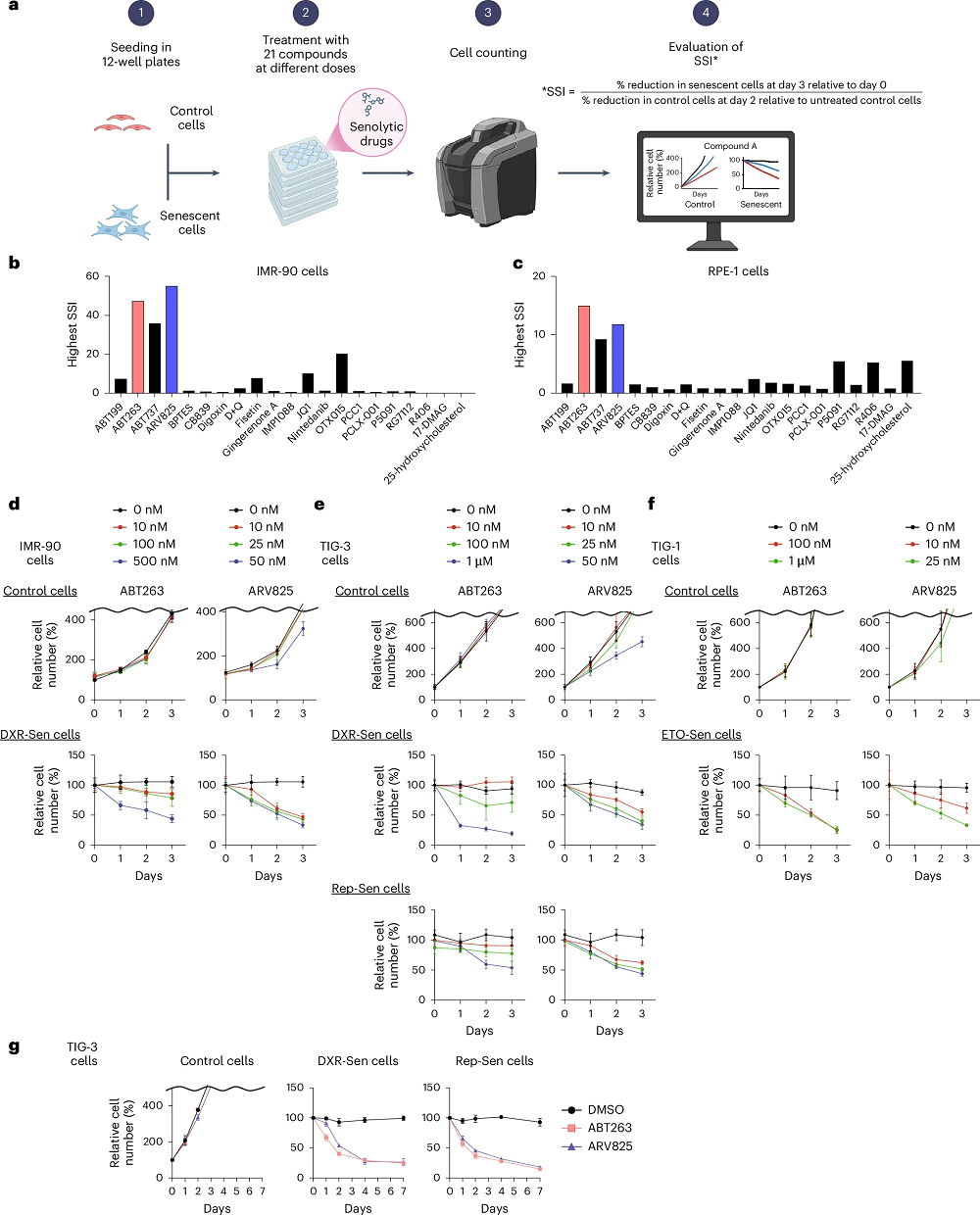
DRRD showed the strongest association with longevity, as the top quintile had 24% lower mortality than the bottom quintile. The authors attribute this to the fact that DRRD’s scoring algorithm directly includes dietary fiber and glycemic index, the two components that individually showed the strongest associations with mortality: fiber was protective, while high glycemic index was detrimental. Product-wise, sugar-sweetened beverages turned out to be the most harmful, in line with previous research [3].
Other scores followed DRRD closely, with 20% reductions in mortality for the top quintiles of AHEI and AMED compared to the bottom ones, 19% for DASH, and 18% for hPDI. Interestingly, noticeable sex-related differences were observed. When the researchers looked at life expectancy, the top performer for men was DRRD, with 3 years gained between the lowest and the highest quintiles, while for women, it was AMED, with 2.3 years. For both sexes, the least effective diet was hPDI (1.9 and 1.5 years, respectively).
How do “longevity genes” factor in?
The team then analyzed PRS’ association with remaining life expectancy, and it turned out to be slightly lower: 1.4 years for men and 1.7 years for women. However, the PRS was split into tertiles, so the researchers compared the top and bottom tertiles. Importantly, the effects of diet and genetics were roughly additive, with DRRD showing the largest combined gains in both sexes. Being both in the top DRRD quintile and in the top PRS tertile was associated with 3.2 years of additional life expectancy for men and 5.5 years for women.
However, the combined estimates are peculiarly noisy, especially for men. For instance, having a top AMED score and a top PRS gives only 1 year for men, which is actually less than either diet alone (2.2) or PRS alone (1.4). This is probably because the combined estimates come from much smaller slices of the cohort, resulting in noise, rather than due to any negative interaction. The women’s combined numbers behave more sensibly and are roughly additive.
Crucially, these estimates are for a 45-year-old person. The life expectancy benefits of switching to a better diet, naturally, diminish with age, as fewer years remain for the risk reduction to play out, while the risk of dying from something unrelated to diet increases.
While this is a well-executed and carefully sensitivity-tested observational study, a few caveats apply. The effect sizes are modest, and the confidence intervals are wide enough for the true benefit to be as small as about 0.5 years in some comparisons. These are also best-case comparisons that compare top and bottom quintiles, while most people usually do not switch their diets from the worst to the best. Finally, the PRS interaction story is intriguing but might not be statistically robust, leaving room for further research.
Literature
[1] Lv, Y., Song, J., Ding, D., Luo, M., He, F. J., Yuan, C., MacGregor, G. A., Liu, L., & Chen, L. (2026). Healthy dietary patterns, longevity genes, and life expectancy: A prospective cohort study. Science advances, 12(7), eads7559.
[2] Afshin, A., Sur, P. J., Fay, K. A., Cornaby, L., Ferrara, G., Salama, J. S., … & Murray, C. J. (2019). Health effects of dietary risks in 195 countries, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. The lancet, 393(10184), 1958-1972.
[3] Imamura, F., O’Connor, L., Ye, Z., Mursu, J., Hayashino, Y., Bhupathiraju, S. N., & Forouhi, N. G. (2015). Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: systematic review, meta-analysis, and estimation of population attributable fraction. Bmj, 351.