The researchers begin this paper with a discussion of well-known interventions and their drawbacks. For example, they note that while rapamycin is effective in extending the lifespans of mice [1], it is associated with cancer development [2]; while adding the anti-cancer drug trametinib mitigates this issue [3], this combination strongly inhibits mTOR, which is necessary for stem cell and brain function [4]. Similarly, knocking out the interleukin IL-11 extends lifespan in mice [5], but its levels only increase with severe disease [6] and it plays a major part in ovarian health [7].
Therefore, these researchers have built on their previous work involving the systemic environment. The Conboy lab is most famous for its work on heterochronic parabiosis and therapeutic plasma exchange, which remove harmful compounds from the bloodstream that accumulate with age [8]. However, such an approach involves repeatedly replacing a person’s blood, which comes with its own complications [9].
This experiment involves attempting to recapitulate the benefits of TPE by addressing its key molecular determinants. This team found two that it deemed likely to work in conjunction: oxytocin, a well-known compound involved in social bonding [10] and is crucial in healing and metabolism [11], and A5i, a compound that inhibits the age-related increase of TGF-β, a factor that promotes fibrosis and inflammation [12]. This lab’s previous work has found that this combination, OT+A5i, leads to tissue rejuvenation [13], and so it performed another experiment involving lifespan.
Sex-dependent effects
The researchers’ lifespan experiment involved four separate groups of males and females either receiving OT+A5i or serving as controls, each of which contained roughly a dozen animals. These mice, at 25 months of age, were already old and frail at the beginning of the study. For two weeks, they would be given this combination three times a week, then left for two weeks without the combination before they were physiologically assessed and the treatment cycle was repeated.
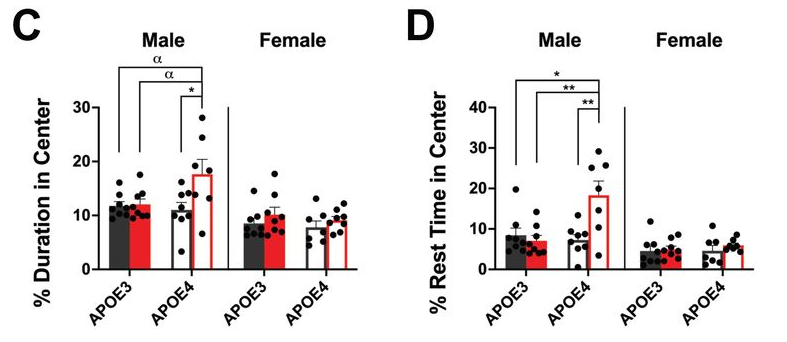
In male mice, the gains in lifespan were tremendous. While their lifespans were only increased by an average of 14% as measured from birth, they were increased by 74% as measured from the beginning of treatment. Six months after treatment, more than three quarters of the treated male mice were still alive, while only about a third of the male mice in the control group had survived this long.
Unfortunately, there were no benefits for lifespan in female mice. The survival curves of the controls and OT+A5i female mice looked similar, with a trend towards a decrease in lifespan instead.
The researchers further assessed this combination’s effects on frailty. They found that OT+A5i improved healthspan as well as lifespan, increasing the time before the animals reached a certain threshold of assessed frailty according to multiple physical measurements, such as vision, gait, and physical challenges such as treadmill and limb hanging times. Additionally, the treated males were more likely to live longer even after becoming frail.
There were clear effects on the individual metrics used to assess frailty as well. The treated male mice were much more able to recognize novel objects, run for longer on gradually accelerating treadmills, and hang for longer from a wire ceiling. Similarly to the lifespan study, none of these beneficial effects on healthspan were found on old female mice.
Only short-term effects on females
These sex-dependent results appear to have been due to a lack of long-term efficacy in females. Immediately after administration of OT+A5i, the researchers discovered that both male and female mice began to synthesize a more youthful balance of proteins (the proteome). Proteomic noise was also, in the short term, decreased by OT+A5i in both sexes. However, these effects were attenuated in females but not males after four months of treatment.
The researchers note that oxytocin is already approved for clinical use by the FDA and that A5i drugs are already being investigated for the treatment of certain conditions, with no major adverse effects yet reported in clinical trials. Therefore, it would be logical to begin a clinical trial to investigate what, if any, effects that this combination may have on the lifespan and healthspan of older men.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
[1] Bjedov, I., & Rallis, C. (2020). The target of rapamycin signalling pathway in ageing and lifespan regulation. Genes, 11(9), 1043.
[2] Bitto, A., Ito, T. K., Pineda, V. V., LeTexier, N. J., Huang, H. Z., Sutlief, E., … & Kaeberlein, M. (2016). Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. elife, 5, e16351.
[3] Gkioni, L., Nespital, T., Baghdadi, M., Monzó, C., Bali, J., Nassr, T., … & Partridge, L. (2025). The geroprotectors trametinib and rapamycin combine additively to extend mouse healthspan and lifespan. Nature Aging, 1-17.
[4] Garza-Lombó, C., Schroder, A., Reyes-Reyes, E. M., & Franco, R. (2018). mTOR/AMPK signaling in the brain: Cell metabolism, proteostasis and survival. Current opinion in toxicology, 8, 102-110.
[5] Widjaja, A. A., Lim, W. W., Viswanathan, S., Chothani, S., Corden, B., Dasan, C. M., … & Cook, S. A. (2024). Inhibition of IL-11 signalling extends mammalian healthspan and lifespan. Nature, 632(8023), 157-165.
[6] Ren, C., Chen, Y., Han, C., Fu, D., & Chen, H. (2014). Plasma interleukin-11 (IL-11) levels have diagnostic and prognostic roles in patients with pancreatic cancer. Tumor Biology, 35(11), 11467-11472.
[7] Cork, B. A., Li, T. C., Warren, M. A., & Laird, S. M. (2001). Interleukin-11 (IL-11) in human endometrium: expression throughout the menstrual cycle and the effects of cytokines on endometrial IL-11 production in vitro. Journal of reproductive immunology, 50(1), 3-17.
[8] Kim, D., Kiprov, D. D., Luellen, C., Lieb, M., Liu, C., Watanabe, E., … & Conboy, I. M. (2022). Old plasma dilution reduces human biological age: a clinical study. Geroscience, 44(6), 2701-2720.
[9] Mokrzycki, M. H., & Kaplan, A. A. (1994). Therapeutic plasma exchange: complications and management. American Journal of Kidney Diseases, 23(6), 817-827.
[10] Zeki, S. (2007). The neurobiology of love. FEBS letters, 581(14), 2575-2579.
[11] Breuil, V., Trojani, M. C., & Ez-Zoubir, A. (2021). Oxytocin and bone: review and perspectives. International journal of molecular sciences, 22(16), 8551.
[12] Tzavlaki, K., & Moustakas, A. (2020). TGF-β Signaling. Biomolecules, 10(3), 487.
[13] Mehdipour, M., Etienne, J., Chen, C. C., Gathwala, R., Rehman, M., Kato, C., … & Conboy, I. M. (2019). Rejuvenation of brain, liver and muscle by simultaneous pharmacological modulation of two signaling determinants, that change in opposite directions with age. Aging (Albany NY), 11(15), 5628.
Date Posted: September 26, 2025
Comments Off on New Universal Therapy Effective in Multiple Tumor Types
Scientists have reported a breakthrough in treating solid tumor cancers using a Velcro-like tool that targets glycans, surface sugars especially abundant in cancer cells. This potentially off-the-shelf therapy does not need adjustment to individual cancer types or patients.
No sugarcoating this
Antibody-based cancer immunotherapies, such as chimeric antigen receptor T (CAR-T) cells were once hailed as gamechangers in oncology. While their potential is indeed massive, several major hurdles remain. For instance, such tools need very high affinity to kill tumors, but that high affinity means they risk hitting the same target on normal tissues, where it can be present in low numbers (“on-target, off-cancer” toxicity) [2]. Moreover, targets tend to be specific to a cancer type or even a particular tumor, requiring expensive tailor-made approaches.
One of the most widespread adaptations that help cancer cells grow and propagate while evading the immune system is a remodeled glycocalyx, which is a cellular coating of glycans linked to proteins and fats (glycoproteins, glycolipids, proteoglycans) [3]. Tumors often thicken and re-pattern these sugars into tumor-associated carbohydrate antigens (TACAs), which can hide danger signals from immune cells, boost growth, and make it physically harder for immune cells to latch on.
However, this glycocalyx remodeling also differentiates cancer cells from the rest and provides an opportunity to target them. If we find a way to target TACAs safely, we can get a single approach that works across many tumors while sparing healthy cells. This idea is at the heart of a new study from the University of California Irvine, published in the journal Cell.
The two-armed bandit
The researchers used sugar-binding proteins called lectins in a way that causes many weak grips to add up (avidity), which the authors liken to Velcro, rather than the single high-affinity “key-lock” connection characteristic of antibodies. They created a bispecific protein that fuses a lectin carbohydrate-recognition domain (CRD) to a single-chain antibody.
Bispecificity means that one arm of the protein, containing four lectins, like four Velcro hooks, latches on to the glycans on the cancer cell’s surface, while the other arm attaches itself to a T cell, bringing it into proximity with the cancer cell. The researchers hypothesized that this construct, glycan-dependent T-cell recruiter (GlyTR, pronounced “glitter”), would recruit T cells to high-TACA-density cancer cells but not to normal cells, where glycan density is much lower.
“It’s the holy grail – one treatment to kill virtually all cancers,” said Michael Demetriou, MD, Ph.D., a professor of neurology, microbiology and molecular genetics at the UC Irvine School of Medicine and the paper’s corresponding author. “GlyTR’s velcro-like sugar-binding technology addresses the two major issues limiting current cancer immunotherapies: distinguishing cancer from normal tissue and cancer’s ability to suppress the immune system.”
The researchers confirmed that the GlyTR’s lectin arm binds to cancer cells but not to the T cells that GlyTR engages. They also tested GlyTRs on several types of healthy tissues and red blood cells (RBCs), confirming there was no strong interaction with non-cancer cells or concerning accumulation in normal organs in vivo.
Effective against multiple cancers
For cancer, however, the effect was devastating. In vitro, both GlyTR versions created by the researchers, GlyTR1 and GlyTR2, bound a wide variety of solid and liquid tumors, including breast, ovarian, prostate, pancreatic, colon, lung, AML, myeloma, and T-cell leukemia, much more than normal lymphocytes and drove T-cell-dependent killing even at very low concentrations. Notably, conventional immunotherapies mostly work for blood cancers, while solid tumors proved to be much harder to crack.
For their in vivo experiments, the team used immunodeficient mice given human T cells (humanized NSG mice) and implanted rejection-proof human tumors. When they injected fluorescent GlyTRs into the bloodstream, the proteins homed to lungs that harbored metastatic triple-negative breast cancer (TNBC) but not to lungs without tumors, providing evidence of tumor-seeking behavior in vivo.
In intraperitoneal models of TNBC, pancreatic, and ovarian cancers, GlyTRs produced dramatic response compared to just human CD8+ T cells, completely blocking tumor growth. The ovarian cancer model was independently replicated at the National Cancer Institute in a good sign for future clinical trials.
Clinical grade GlyTR1 protein manufacturing is already being developed at the NCI Experimental Therapeutics program labs in Maryland. Due to these phenomenal results, the team plans to bring the project to the clinic as early as possible, with human trials in about two years from now, according to the press release.
“This is the revolutionary approach to cancer treatment our patients have been waiting for,” said Farshid Dayyani, MD, Ph.D., medical director of the Stern Center for Clinical Trials and Research. “We are committing all available resources to bring this exciting new trial to UCI Health as fast as possible.”
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
[1] Zhou, R. W., Purohit, P. K., Kim, J. H., Newton, B. L., Edwards, R. A., & Demetriou, M. (2025). Safe immunosuppression-resistant pan-cancer immunotherapeutics by velcro-like density-dependent targeting of tumor-associated carbohydrate antigens. Cell, 188, 1–17.
[2] Lamers, C. H., Sleijfer, S., Van Steenbergen, S., Van Elzakker, P., Van Krimpen, B., Groot, C., … & Gratama, J. W. (2013). Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Molecular therapy, 21(4), 904-912.
[3] Buffone Jr, A., & Weaver, V. M. (2019). Don’t sugarcoat it: How glycocalyx composition influences cancer progression. Journal of Cell Biology, 219(1), e201910070.
Date Posted: September 25, 2025
Comments Off on A Potential Reason Why Clotting Increases With Age
It is well known that blood clots, which form when platelets bind together, are a serious problem in older people [1]. Arterial clots directly cause both heart attacks and strokes, often being the final capstones on gradual cardiovascular aging, but changes in HSC behavior drive many other conditions as well [2].
In the canonical pathway, HSCs go through three intermediate cell types before finally becoming megakaryocyte progenitors (MkPs), which form the megakaryocytes that release platelets [3]. This team has previously discovered that a non-canonical pathway also exists and increases with age, turning HSCs directly into MkPs with no intermediate steps [4]. Worse, this previous work found that platelets created by non-canonical MkPs (ncMkPs) are hyperactive, heightening their potential contributions to clotting-related diseases.
To illustrate these differences, the researchers had created a mouse model, FlkSwitch, that expresses green fluorescent protein (GFP) in MkPs that go through the canonical pathway and a different fluorescent protein, Tomato, in MkPs that go through the faster, non-canonical pathway. This paper is a closer examination into the differences between these MkPs, as the researchers wanted to be able to identify them without the need for specialized mice and to determine the functional differences between them.
Finding the right biomarkers
In their first experiment, the researchers combined their previous bulk-sequencing RNA results with single-cell sequencing. They were looking for membrane-bound proteins, which could potentially be used in future studies or even clinical practice to determine which MkPs are which, and they sought proteins for which commercially available antibodies already exist.
Initially, the researchers hit upon CD54 as their target protein, but further work found that CD48 was a better fit; MkPs expressing more CD48 are much more likely to be created through the canonical pathway, as determined by GFP and Tomato comparisons. Another protein, CD321, was found to be a strong predictor in the other direction; MkPs that express high amounts of this protein are much more likely to be created through the non-canonical pathway. This combination of CD48 and CD321 was highly useful in creating a firm basis for differentiating MkPs, as it yielded similar results to those gleaned from the FlkSwitch mice.
A further experiment found that ncMkPs are not entirely exclusive to aged animals. In 2- to 3-month-old male mice, approximately one in five MkPs is created through the non-canonical pathway. In aged male mice, however, this changes to an average of three in five, although there are large variations between animals. For female mice, there was a statistically significant increase with aging, but it was somewhat less noticeable and older female mice have fewer platelets overall than older male mice do, despite having similar numbers of MkPs.
Hardier survivors
One crucial and surprising finding was that older ncMkPs have a much greater ability to survive in vitro than all other MkPs. When derived from younger animals, ncMkPs proliferate poorly, less than canonical MkPs (cMkPs) of any age. Older ncMkPs, on the other hand, proliferate much more.and are much less likely to die by apoptosis.
This increase in activity persisted in vivo as well; older ncMkPs that were given to younger mice, which had their relevant cells previously destroyed by radiation, proliferated much more than younger ncMkPs did. Additionally, chemical stimulation determined that the platelets produced by older ncMkPs were, indeed, extremely reactive.
The environment plays a role
Taking HSCs from younger and older mice and observing their conversion into MkPs yielded entirely expected results; HSCs derived from the older animals were much more likely to produce ncMkPs. However, these findings changed when these cells were introduced into a younger irradiated animal; there, HSCs produced similar proportions of MkPs, regardless of whether they had come from a younger or older source. A closer examination to determine if any individual HSC is more or less likely to prouce ncMkPs yielded no useful results, suggesting that the aged environment is a strong factor in HSCs creating ncMkPs
As the researchers point out, however, humans and mice differ, and platelet generation is one of those differences; in humans, platelet count decreases with age [5]. Therefore, it is not clear if these murine findings could directly translate to human beings. However, with the identification of the proteins CD48 and CD321 as biomarkers, further work on human MkPs may reveal whether this change in cell fate is a significant factor in human clot-related diseases along with potential interventions to address it.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
[1] Le Blanc, J., & Lordkipanidzé, M. (2019). Platelet function in aging. Frontiers in cardiovascular medicine, 6, 109.
[2] Chung, S. S., & Park, C. Y. (2017). Aging, hematopoiesis, and the myelodysplastic syndromes. Blood advances, 1(26), 2572-2578.
[3] Manso, B. A., Rodriguez y Baena, A., & Forsberg, E. C. (2024). From hematopoietic stem cells to platelets: Unifying differentiation pathways identified by lineage tracing mouse models. Cells, 13(8), 704.
[4] Poscablo, D. M., Worthington, A. K., Smith-Berdan, S., Rommel, M. G., Manso, B. A., Adili, R., … & Forsberg, E. C. (2024). An age-progressive platelet differentiation path from hematopoietic stem cells causes exacerbated thrombosis. Cell, 187(12), 3090-3107.
[5] Jones, C. I. (2016). Platelet function and ageing. Mammalian genome, 27(7), 358-366.
Date Posted: September 24, 2025
Comments Off on Personalized Medicine Approach to Senolytics Clinical Trials
Destroying senescent cells using senolytics may mitigate some aging-associated symptoms or diseases. Preclinical studies on senolytics have determined that they can induce many beneficial effects, thus prompting researchers to test them against various conditions in human clinical trials. Those studies have determined that senolytics appear to be safe for use in people.
The authors of this commentary summarized the results of those clinical trials, dividing them into two groups. The first encompassed trials that used senolytics as systemic treatments, and the second encompassed trials in which senolytics were used locally due to possible toxicity. The discussion mainly focused on the systemic treatment trials.
Of the systemic treatments, only the combination of dasatinib and quercetin has been tested; those trials included a small number of participants, with feasibility and safety being the primary focus. While some trials suggest efficacy, the authors caution against drawing firm conclusions since some studies didn’t have control groups to compare against.
Systemic senolytic treatment was also used in two randomized controlled trials. One of them was published by the same group that wrote this commentary, and it involved the most extensive study of this type with 60 postmenopausal women [2]. The researchers observed “a positive signal for an increase” in the bone formation marker procollagen type 1 N-propeptide (P1NP). They note that analyzing this study suggested some improvements that can be applied in the future design of senolytics trials. To explain this, they dived deeper into the results.
Assessing senescent cell burden
First, they describe that when analyzing the entire study population, they observed an increase in the bone formation marker P1NP, but it was modest (16% increase compared to controls). However, the study hypothesized that “the baseline senescent cell burden would determine the clinical response to the senolytic intervention.” To test that hypothesis, the researchers needed to assess senescence burden by measuring the gene expression of p16 in T cells. However, since such measurements are challenging, they also conducted additional tests and measured a panel of 36 SASP factors.
When women were divided into groups based on T cell expression of p16 mRNA, the women in the highest tertile (T3) had the highest P1NP levels. They also identified six SASP factors that showed increased levels at baseline in the T3 group compared to the T1 and T2 groups. They used those six factors to develop a SASP score that indicated senescent cell burden and used this score to predict responses to dasatinib and quercetin treatment: the SASP-score T3 group showed a similar increase in P1NP as the T cell p16 T3 group. Moreover, selecting participants in both the SASP-score T3 group and the T cell p16 T3 group (the people with the highest senescent cell burden) showed even higher levels of P1NP following dasatinib and quercetin treatment, supporting the original hypothesis.
The authors note that while the levels of p16 in T cells appear to be helpful in grouping patients in clinical trials, there is a need to understand the underlying biology behind this connection and assess the variability of p16 levels over time and their correlation with the SASP.
The authors hypothesize that the relationship between senescent cell burden and senolytics might be even more complex. They elaborate that senescent cell burden increases as we age and during diseases. They suggest that the first-generation senolytics, such as dasatinib and quercetin, which have modest potency, are effective in people with high senescent cell burdens, and they hypothesize that second-generation, more potent senolytics (currently under development) should also be effective in people with lower senescent cell burdens. Their hypothesis should be tested once those compounds are developed.
Developing resistance
The authors also point to another unresolved issue that their study identified. They observed the peak of increase in the bone formation marker serum P1NP at week four after starting the dasatinib plus quercetin treatment, but later those levels returned to baseline. One possible explanation can be an induction of compensatory mechanisms. However, the authors also suggest an alternative explanation. They suggest that after initial removal of a subset of senescent cells, remaining cells develop resistance to senolytics. Such a mechanism has never been reported in clinical trials; however, they believe it needs consideration while designing future senolytics clinical trials. The authors suggest two possible approaches that can be applied in future trials to address such an issue: either extending the time between dosing to prevent the development of resistance or incorporating combined treatment with another senolytic that targets different molecular pathways.
The personalized medicine of the future
The authors’ observations warrant further investigation into personalized approaches during senolytics treatment. Since multiple mechanisms drive aging, it is essential to identify the people for whom senescence is a major factor in aging, such as people with high senescent cell burdens, as they might benefit the most from senolytics treatment.
Further, the authors recommend future research that focuses on developing improved biomarkers of senescent cell burden. They believe that the currently used measurement of T cell p16 expression levels or the SASP score they developed, which should still be validated for outcomes unrelated to the skeleton, should be expanded. They propose using large-scale omics-based approaches to develop organ-specific biomarkers for senescent cell burden, which would aid in selecting clinical trial participants with a high senescent cell burden in any particular study’s relevant organ.
In summary, the authors recommend that future clinical trials of senolyics focus on better biomarkers of senescent cell burden and test whether individuals identified with those markers as having high senescent cell burdens indeed respond best to senotherapeutic therapies.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
[1] Khosla, S., Monroe, D. G., & Farr, J. N. (2025). Towards a personalized approach in senolytic trials. Nature aging, 10.1038/s43587-025-00964-5. Advance online publication.
[2] Farr, J. N., Atkinson, E. J., Achenbach, S. J., Volkman, T. L., Tweed, A. J., Vos, S. J., Ruan, M., Sfeir, J., Drake, M. T., Saul, D., Doolittle, M. L., Bancos, I., Yu, K., Tchkonia, T., LeBrasseur, N. K., Kirkland, J. L., Monroe, D. G., & Khosla, S. (2024). Effects of intermittent senolytic therapy on bone metabolism in postmenopausal women: a phase 2 randomized controlled trial. Nature medicine, 30(9), 2605–2612.
Date Posted: September 23, 2025
Comments Off on Exercise Suppresses Appetite via a Brain Pathway
It has been long known that, somewhat counterintuitively, exercise transiently suppresses appetite. Scientists suspect that this contributes to exercise-related weight loss. However, the exact mechanisms behind this effect, which can possibly be used to help treat obesity and metabolic disorders, were not well understood. In this new study published in Nature Metabolism, researchers from Baylor College of Medicine, Jan and Dan Duncan Neurological Research Institute (Duncan NRI) at Texas Children’s Hospital, Stanford University School of Medicine, and collaborating institutions, reveal one such mechanism that works through the brain.
“Regular exercise is considered a powerful way to lose weight and to protect from obesity-associated diseases, such as diabetes or heart conditions,” said co-corresponding author Dr. Yang He, assistant professor of pediatrics – neurology at Baylor and investigator at the Duncan NRI. “Exercise helps lose weight by increasing the amount of energy the body uses; however, it is likely that other mechanisms are also involved.”
The two-stage neuronal pathway
Prior work demonstrated that intense exercise raises blood levels of N-lactoyl-phenylalanine (Lac-Phe, a small molecule made from lactate and the amino acid phenylalanine) and that dosing mice with Lac-Phe suppresses appetite without obvious side effects [2]. Another study found that Lac-Phe gets elevated by the anti-diabetes drug metformin [3]. The researchers of this new study set out to answer which brain cells Lac-Phe acts on to curb feeding.
First, they reconfirmed its basic effect by injecting Lac-Phe into the abdomen and brain ventricles of mice. This resulted in reduced food intake in both normal-diet and high-fat-diet mice, suggesting a brain-based mechanism of action rather than something related to gut distress. They also ran behavioral tests to show that the compound wasn’t just making mice feel sick.
The team then stained brains for c-Fos, a protein used as a marker of recently activated neurons, after Lac-Phe administration. Two regions showed increased activity: the nucleus tractus solitarius (NTS) and the paraventricular nucleus of the hypothalamus (PVH). This marked both as potential relay points.
The researchers then selectively silenced the exact NTS neurons or PVH neurons that Lac-Phe had activated. Silencing the NTS neurons did not change appetite suppression by Lac-Phe, while silencing the PVH neurons blunted it. This suggested that PVH activation is required for feeding suppression, while NTS activation is incidental.
However, PVH neurons turned out to be the downstream part of the pathway. Experiments showed that a large fraction of their input comes from Agouti-related peptide (AgRP) neurons in the arcuate nucleus (ARH). These neurons are classic “hunger” cells, well known for their role in appetite regulation through AgRP, neuropeptide-Y (NPY), and the neurotransmitter GABA. Indeed, applying AgRP or NPY directly inhibited PVH neurons.
Direct application of Lac-Phe to AgRP neurons dose-dependently reduced their firing, and a set of further experiments confirmed their role. This suggested that when Lac-Phe inhibits AgRP neurons, PVH neurons become disinhibited, causing animals to eat less. The researchers then showed that the post-exercise dip in feeding depended on the inhibition of AgRP neurons.
“Understanding how Lac-Phe works is important for developing it or similar compounds into treatments that may help people lose weight,” He said. “We looked into the brain as it regulates appetite and feeding behaviors.”
Fight or flight, but don’t eat
The researchers were also able to show that Lac-Phe directly quiets AgRG hunger neurons by opening KATP: energy-sensing potassium channels on cell membranes that inhibit neuronal activity when energy is plentiful.
“We found that Lac-Phe acts on a protein on AgRP neurons called the KATP channel, which helps regulate cell activity. When Lac-Phe activates these channels in AgRP neurons, the cells become less active,” Dr. He said. “When we blocked the KATP channels using drugs or genetic tools, Lac-Phe no longer suppressed appetite. This confirmed that the KATP channel is essential for Lac-Phe’s effects.”
These results suggest a specific ion-channel mechanism that can be targeted by future therapies to recapitulate the weight loss associated with exercise. “This finding is important because it helps explain how a naturally produced molecule can influence appetite by interacting with a key brain region that regulates hunger and body weight,” said co-corresponding author Dr. Jonathan Long at Stanford University School of Medicine.
Interestingly, according to the researchers, previous studies have shown that Lac-Phe is most elevated by sprinting, followed by resistance training and then endurance training. There might be a deep evolutionary reason here: if you have to run, it is possible that this is not an isolated event but rather a sign that you are in a dangerous environment that might require more running. Keeping appetite down for a while should make future sprinting easier.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
[1] Liu, H., Li, V. L., Liu, Q., Liu, Y., Su, C., Wong, H., … & Xu, Y. (2025). Lac-Phe induces hypophagia by inhibiting AgRP neurons in mice. Nature Metabolism, 1-14.
[2] Li, V. L., He, Y., Contrepois, K., Liu, H., Kim, J. T., Wiggenhorn, A. L., … & Long, J. Z. (2022). An exercise-inducible metabolite that suppresses feeding and obesity. Nature, 606(7915), 785-790.
[3] Xiao, S., Li, V. L., Lyu, X., Chen, X., Wei, W., Abbasi, F., … & Long, J. Z. (2024). Lac-Phe mediates the effects of metformin on food intake and body weight. Nature metabolism, 6(4), 659-669.
Date Posted: September 22, 2025
Comments Off on AI Model Accurately Predicts Multiple Disease Risks
For millennia, people have wanted to know what health events await them in the future. Models have been created that can forecast the onset of a single disease reasonably well. However, predicting several health outcomes simultaneously has been proven tricky. According to a new study published in Nature by an international group of researchers the immense power of AI can be harnessed to solve this problem.
The researchers created a model based on the GPT architecture, which powers chatbots such as ChatGPT. The model was fed data from UK Biobank, a huge repository of longitudinal health data on some half of a million British citizens.
The team notes that health events, biomarkers, and risk factors create an interconnected network that somewhat resembles language. Just like a large language model, such as GPT, predicts the next word, a model trained on health data can predict the next outcome. Age, sex, BMI, and risk factors such as tobacco use were also included in the model.
“Here we demonstrate,” the paper says, “that attention-based transformer models, similar to LLMs, can be extended to learn lifetime health trajectories and accurately predict future disease rates for more than 1,000 diseases simultaneously on the basis of previous health diagnoses, lifestyle factors and further informative data.”
Big results from big data
The model accepts the patient’s previous health history as a prompt. It then predicts the probability of the next health event in their life (for example, “pneumonia 18%, heart failure 9%, death 3%”, and so on) and the time to that event, with accuracy comparable to that of single-disease tools. Consequently, it can “generate entire future health trajectories,” study co-author Moritz Gerstung, a data scientist at the German Cancer Research Center in Heidelberg, said to Nature. “A health care professional would have to run dozens of them to deliver a comprehensive answer,” he added.
The model, called Delphi-2M (after the two million parameters it uses), also generally outperformed a multi-disease predictor trained on 67 UK Biobank biomarkers. However, its predictive power trailed some strong lab markers, such as HbA1c for diabetes, underscoring the value of biomarkers for some endpoints.
The model was validated and tested on UK Biobank data that was not used for training. Then, it was also tested on a separate Danish dataset of about 1.9 million health trajectories, where it showed only slightly reduced accuracy.
Delphi-2M can be used not only for flagging potential health concerns in individuals but also for populational modeling. For instance, it can model thousands of health trajectories for a given region and demographic mix, producing forward-looking estimates of incidence, hospitalizations, deaths, and years lived with disease. Because it preserves competing risks, it also enables ‘what-if’ analysis; for instance, it can estimate the gain in average life expectancy from eliminating cancer.
Delphi-2M, named after the legendary Greek oracle, cannot actually predict death with complete certainty. It can, by simulating many futures, draw a survival curve, showing how your risk of death changes with age. This is simply risk stratification and planning, with uncertainty bands and competing risks baked in, rather than fortune telling.
The data bottleneck
The researchers acknowledge several potential problems with their design. One is related to the data they used: UK Biobank recruits people aged 40-70. By this age, some people have already died, and the absence of their health trajectories from the data creates bias. The follow-up is limited in time, and hence, it misses people older than 80, meaning that very old-age dynamics are not represented.
Scarcity of data is a major bottleneck for most large AI models in biology. Health systems in various countries sit on mountains of health data spanning from birth to death. Figuring out ways to free this data, while addressing safety concerns and maintaining anonymity, can help accelerate progress in this area. Data from wearables, which are becoming increasingly popular, is another promising source.
Yet another way to augment data for large models is by using synthetic data: that is, data simulated by the model itself. Delphi-2M was asked to generate fake patient histories, and then a new model was trained only on those synthetic records without any real people’s data. Surprisingly, the model trained exclusively on synthetic data was almost as accurate in its predictions as the original model that used UK Biobank data. This approach can help solve the anonymity problem by minimizing the use of real patients’ data.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
[1] Shmatko, A., Jung, A. W., Gaurav, K., Brunak, S., Mortensen, L. H., Birney, E., … & Gerstung, M. (2025). Learning the natural history of human disease with generative transformers. Nature, 1-9.
Date Posted: September 19, 2025
Comments Off on Lipid Metabolism Is Causal in Some Alzheimer’s Cases
The key characteristics of Alzheimer’s disease, such as cognitive decline and brain deterioration, are very well-known [1]. However, other symptoms, such as pain sensitivity, may precede these key manifestations, providing an early warning of its development [2].
Previous research has pinpointed lipid metabolism as a key aspect of the relationship between pain and Alzheimer’s [3]. Changes in these lipids have been found to be among the first signs of Alzheimer’s, and they contribute to damage and inflammation [4]. In particular, lysophosphatidylcholine (LPC) is strongly associated with pain [5] along with both neuroinflammation and the removal of protective myelin from axons (demyelination) [6].
However, there are confounding factors in this relationship. Previous work has found that the allele APOE4, which greatly increases sensitivity to Alzheimer’s, plays a role [7]. Alzheimer’s presentation and pain sensitivity also vary by sex [8].
The biological chain of causality between these facts, however, had not been examined. These researchers aimed to rectify that by taking a close look at LPC acyltransferase 2 (LPCAT2), a core part of lipid metabolism and brain inflammation.
Large databases help find a limited group
Because of the number of involved variables, the researchers needed to use multiple large databases: the Alzheimer’s Disease Neuroimaging Initiative, the ROSMAP database on memory and aging, a Mayo Clinic database on gene expression in the brain, and the Taiwan Biobank were all used as data sources along with human brain samples from the NIH.
The researchers discovered that pain sensitivity is increased with mild cognitive impairment in men that do not have APOE4. Women, and men with APOE4, did not have results that reached the level of statistical significance. In fact, women without APOE4 trended towards suffering less pain with cognitive decline as measured by the MMSE, a commonly used metric of cognitive ability.
A gene expression analysis found that this increase in pain sensitivity in non-APOE4 men was indeed related to LPCAT2, which was associated with both increased pain and with the onset of Alzheimer’s disease. Men who did not express increased levels of LPCAT2 were unlikely to experience dementia; men who did had a much greater risk.
These findings were corroborated with an analysis of brain tissue. Nearly every sample that was derived from a non-APOE4 man with Alzheimer’s disease contained elevated LPCAT2. This was even found to be true in mice; male mice that were modified to be susceptible to Alzheimer’s disease were considerably more likely to have elevated LPCAT2 in the hippocampus, although this finding did not extend to the cerebral cortex.
Genetic susceptibility
The researchers found that there is a genetic component. Eleven single-nucleotide polymorphisms (SNPs) were found to be associated with increased LPCAT2, pain sensitivity, and the risk of Alzheimer’s disease. Mendelian randomization, a statistical technique, was used to confirm that this relationship is causal; these mutations do indeed raise the risk of increased pain sensitivity and Alzheimer’s disease in non-APOE4 men.
The researchers offer some hypotheses as to why this might be the case. They note that APOE is directly related to circulating LPC levels [9] and microglial behavior [10] and that estrogen has been found to play a role in this area as well [11]. However, this study is observational and does not suggest any methods of modulating LPCAT2. Determining whether or not this is a druggable target will be the domain of future work.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
[1] Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., & Jones, E. (2011). Alzheimer’s disease. the Lancet, 377(9770), 1019-1031.
[2] Zhao, W., Zhao, L., Chang, X., Lu, X., & Tu, Y. (2023). Elevated dementia risk, cognitive decline, and hippocampal atrophy in multisite chronic pain. Proceedings of the National Academy of Sciences, 120(9), e2215192120.
[3] Yin, F. (2023). Lipid metabolism and Alzheimer’s disease: clinical evidence, mechanistic link and therapeutic promise. The FEBS journal, 290(6), 1420-1453.
[4] Bazan, N. G., Colangelo, V., & Lukiw, W. J. (2002). Prostaglandins and other lipid mediators in Alzheimer’s disease. Prostaglandins & other lipid mediators, 68, 197-210.
[5] Ren, J., Lin, J., Yu, L., & Yan, M. (2022). Lysophosphatidylcholine: Potential target for the treatment of chronic pain. International Journal of Molecular Sciences, 23(15), 8274.
[6] Freeman, L., Guo, H., David, C. N., Brickey, W. J., Jha, S., & Ting, J. P. Y. (2017). NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. Journal of Experimental Medicine, 214(5), 1351-1370.
[7] Romano, R. R., Carter, M. A., Dietrich, M. S., Cowan, R. L., Bruehl, S. P., & Monroe, T. B. (2021). Could altered evoked pain responsiveness be a phenotypic biomarker for Alzheimer’s disease risk? A cross-sectional analysis of cognitively healthy individuals. Journal of Alzheimer’s Disease, 79(3), 1227-1233.
[8] Aggarwal, N. T., & Mielke, M. M. (2023). Sex differences in Alzheimer’s disease. Neurologic clinics, 41(2), 343.
[9] Law, S. H., Chan, H. C., Ke, G. M., Kamatam, S., Marathe, G. K., Ponnusamy, V. K., & Ke, L. Y. (2023). Untargeted lipidomic profiling reveals lysophosphatidylcholine and ceramide as atherosclerotic risk factors in apolipoprotein E knockout mice. International Journal of Molecular Sciences, 24(8), 6956.
[10] Yamamoto, S., Hashidate-Yoshida, T., Yoshinari, Y., Shimizu, T., & Shindou, H. (2024). Macrophage/microglia-producing transient increase of platelet-activating factor is involved in neuropathic pain. Iscience, 27(4).
[11] Karpuzoglu-Sahin, E., Zhi-Jun, Y., Lengi, A., Sriranganathan, N., & Ahmed, S. A. (2001). Effects of long-term estrogen treatment on IFN-γ, IL-2 and IL-4 gene expression and protein synthesis in spleen and thymus of normal C57BL/6 mice. Cytokine, 14(4), 208-217.
Date Posted: September 18, 2025
Comments Off on Looking Back at Summer, Looking Forward to Growth
For those of us in the Northern Hemisphere, autumn is underway. The fall is a time when the leaves that are green turn to brown, so let us see what the Lifespan team has been working on to help our field keep our own metaphorical leaves green and healthy.
Top longevity news stories of Summer 2025
As always, we have been bringing you the best longevity and aging research news this summer; let’s take a look at some of the top stories.
Tragedy at RAADFest
Two people nearly died, and several more sought treatment, after receiving peptide injections at the last RAADfest in Las Vegas. This highlighted some well-known problems and contradictions in the longevity field.
On one hand, there is a sense of urgency that pushes some people to offer and others to try therapies that have not been rigorously tested for safety and/or efficacy. Many people rightfully lament the increasingly lengthy and expensive regulatory process, which is not properly suited for rejuvenating therapies. There is a growing push to extend the “right to try” to all patients and all reasonably safe therapies, and to accelerate clinical trials by relaxing certain regulations and transitioning towards cost-effective methods, such as human organoids and in silico models.
On the other hand, a legitimate concern exists that the longevity movement might be overshadowed, compromised, and ultimately impeded by the murky wave of overblown claims, unfounded promises, unproven therapies, and flat-out “snake oil” cures. To make some sense of all this in the context of the RAADfest incident, we asked several prominent figures in the field about this incident and what led up to it.
A vertical longevity village in San Francisco
In the heart of San Francisco, a tower in the downtown area is transforming into a center for longevity, artificial intelligence, cryptocurrency, and robotics.
In March, three young German business owners, Jakob Drzazga, Christian Nagel, and Christian Peters, acquired a 16-story building located in San Francisco’s Mid-Market district. They plan to convert the tower into a premier collaborative workspace for various innovative fields, including cryptocurrency, artificial intelligence, robotics, and longevity research.
Lifespan journalist Arkadi Mazin attended their inaugural longevity conference in June to cover these developments. Join us to find out what he learned at Viva Frontier Tower.
Rejuve.AI: A new app focused on longevity
Rejuve.AI is a company founded by Jasmine Smith and well known AI researcher Ben Goertzel. Recently, they have launched a new longevity app, and Arkadi caught up with them to find out more.
Apparently, it is much more than simply yet another health and lifestyle app. The founders say it focuses on using real human data to study lifespan and potentially the efficacy of inventions that target aging.
In this interview we also discuss Ben and Jasmine’s predictions on the future for artificial intelligence, aging research, and how to create a world where medical data is controlled by patients.
The Longevity Summit Dublin has become quite the go-to event in the longevity community. The event first launched in 2022 and has grown in popularity since then.
Dublin is most famous for its Guinness brewery along with the pubs and restaurants of Temple Bar and its long history, but there is a significant buzz about longevity there too.
Hosted at Trinity College in the centre of Dublin, the event is very much in the heart of this vibrant city. The event saw a range of great speakers and workshops focused on aging research and advocacy. It really is great to see the popularity of this event growing and including a free public open day.
Lifespan Editor-in-Chief Steve Hill was at the Summit to bring you the highlights from the 2025 Longevity Summit Dublin. Join us as he shares his thoughts on some of the most interesting news from the conference.
Announcing the Lifespan Alliance
We have also launched the Lifespan Alliance, our corporate sponsorship program that brings together purpose-driven businesses and innovative organizations focused on promoting a longer, healthier human lifespan.
Sponsorship from Alliance members allows us to conduct innovative research, reach millions via news and other media, and link prominent figures in science, policy, and technology to speed up advancements.
If you are interested in supporting us as a company, including the benefits of doing so, please consider becoming a member of the Lifespan Alliance.
We would like to thank all of the organizations that have stepped up to join us so far. Your support means the world to us and allows us to continue our important research, advocacy, journalism, and educational activities.
Organizational leadership changes
There have been some changes to leadership at LRI. Keith Comito and Dr. Oliver Medvedik have become the Chief Executive Officer and Chief Scientific Officer, respectively. They will spearhead LRI leadership and enhance the Institute’s outreach and scientific efforts.
These new roles demonstrate LRI’s dedication to merging innovative leadership with scientific precision and utilizing years of experience in ecosystem development to establish a network that can effectively identify and address key challenges in aging research.
A new lease on life for the Rejuvenation Roadmap
The Rejuvenation Roadmap is a database tracking the many interventions against aging currently being developed and tested. It holds information about each therapy’s progress, from initial drug discovery through to clinical trials. It is a one-stop shop for finding out where the field is in bringing aging under medical control.
With that in mind, we are delighted to announce that Michael Rae is now curating the Rejuvenation Roadmap. Michael is the co-author of the well known rejuvenation biotechnology book Ending Aging, was a science writer for SENS Research Foundation, and is an active member of the longevity community.
We have also added two new aging damage types to the Roadmap: extracellular matrix damage and extracellular aggregates. We have added them to the Roadmap as they are important to our research and more accurately encompass our work.
Extracellular matrix damage involves the degradation and restructuring of its structural and functional elements that make up the extracellular matrix. This is the non-cellular part of tissues that offers structural support and a framework for cells.
Extracellular aggregates refer to a type of defective proteins that have lost their functionality. Instead, they have transformed into adhesive, misshapen forms that adhere to the surfaces of our cells and tissues, disrupting their proper functioning.
Michael has already been busy updating the Roadmap with new drugs and interventions. Keep your eyes out for more in the coming months as the database continues to grow.
A new look for our website
You may have noticed our website has changed a bit recently. Our regulars will recall that LEAF and SRF merged in October 2024 to create the Lifespan Research Foundation (LRI). It has taken a while, but we have now merged both org websites into one.
We like to think of our new site as a longevity switchboard, a place to connect with all of our initiatives. You can find the latest news about the field, learn about our exciting research projects at our Mountain View research centre, check out our educational programs, join the Longevity Investor Network, and much more!
Our goal is simple. Everything you need to know about rejuvenation biotechnology in one place. For those of you who visit us for the latest longevity news, please bookmark the news landing page.
Talking cellular senescence at Longevity Summit Dublin
At the 2025 Longevity Summit Dublin in June, Dr. Amit Sharma gave a talk about some of the work happening at our research center. Our Sharma Lab in Mountain View, California is conducting important research on senescent cells and therapeutic approaches to removing them.
Normally, cells becoming senescent is a helpful safety mechanism that helps us to avoid cancer. Damaged and worn-out cells pose a risk and need to be destroyed via a self-destruct process known as apoptosis.
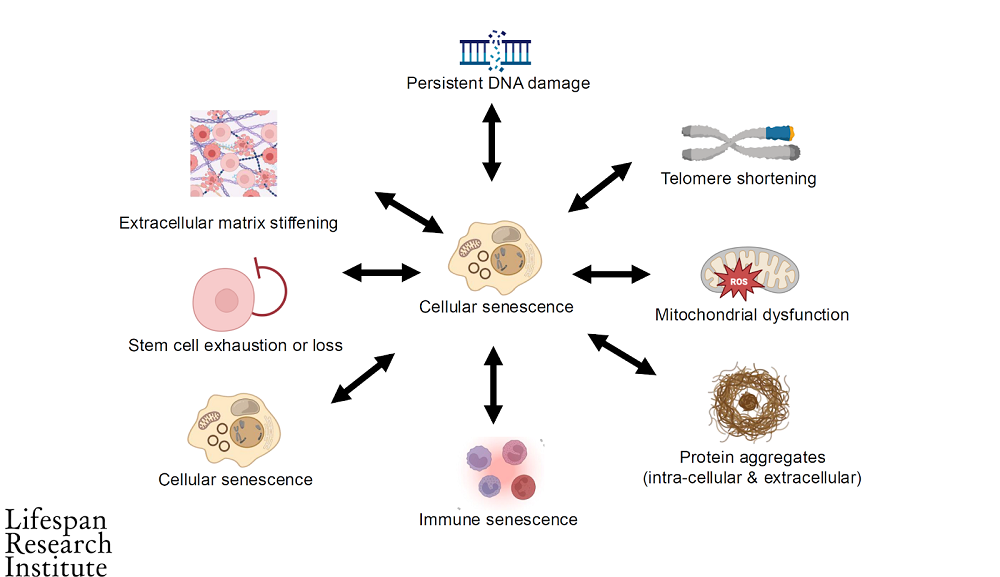
The problem is that as we age, this safety system breaks down and cells that should be removed no longer are. They instead resist apoptosis and remain at large in the body, causing chronic inflammation. The accumulation of excessive numbers of senescent cells is one of the hallmarks of aging.
It has also been determined to affect the other reasons we age. This diagram shows how it affects other age-related damages. Senescent cells appear twice in the image because they can also encourage other nearby healthy cells to become senescent as well. This is caused by the inflammatory secretions known as the senescence-associated secretory phenotype (SASP).
Senescent cell accumulation is linked to a wide variety of age-related diseases:
Cognitive decline
Frailty
Type 2 Diabetes
Obesity-induced dysfunction
Atherosclerosis
Non-alcoholic fatty liver disease (NAFLD)
Alzheimer’s disease
Parkinson’s disease
Idiopathic Pulmonary Fibrosis (IPF)
Chronic obstructive pulmonary disease (COPD)
Liver fibrosis
Osteoarthritis
Osteoporosis
Age-related muscle loss (sarcopenia)
Heart failure
Hypertension
Therapy-induced senescence (TIS)
Rheumatoid arthritis
Kidney disease (Glomerulosclerosis)
Macular degeneration
Chemotherapy-induced fatigue
The research we are doing at the Sharma Lab aims to tackle such diseases by targeting senescent cells. Potentially, this means that therapies based on our research might address many diseases of aging by attacking a core reason that they develop in the first place. Here’s some of the research we are doing at our Mountain View research center.
Iron dyshomeostasis and ferroptosis-based senolytics
Most studies on senescent cells have focused on primary senescent cells. These are the classic senescent cells that become this way due to DNA damage, replicative exhaustion, or mitochondrial dysfunction.
However, more recently, researchers have discovered what are known as paracrine senescent cells, which become senescent due to prolonged exposure to the SASP. This creates a vicious cycle where more and more cells become senescent due to SASP exposure.
Paracrine senescent cells play a key role in the buildup of senescent cells as we age. Previous attempts to define paracrine senescence have been inconsistent because they relied on analyzing mixed groups of both senescent and non-senescent cells.
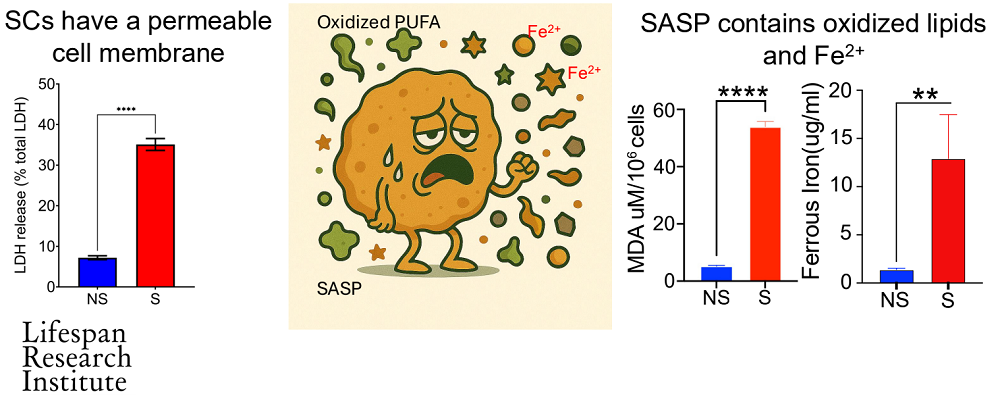
Senescent cells are not destroyed by accumulated iron like healthy cells are. They survive by repairing the plasma membrane and reducing the lipid peroxides induced by iron accumulation. These lipid peroxides create reactive oxygen species (ROS), which the senescent cells actively remove in order to survive.
With this in mind, the team used dipeptidyl peptidase-4 (DPP4) as a surface maker to isolate senescent cells from regular cells. Our researchers found that ferroptosis dysregulation and ferrous iron accumulation are a common phenomenon in both primary and paracrine senescent cells.
Finally, our researchers identified drugs capable of inducing ferroptosis in target senescent cells. Essentially, the cells were already primed for ferroptosis cell death but were resisting it, and these drugs effectively tipped them over the edge. The approach works in primary and paracrine senescent cells.
These results have given us important insights into the nature of senescent cell populations and how we might approach their identification and clearance in a new way.
LAMP1 is a universal surface marker of senescence
Researchers at LRI have identified a cell surface marker of senescence that could make identifying senescent cell populations more reliable.
There have been many attempts to create accurate biomarkers in order to identify senescent cell populations. While there are some methods to do so, these have limitations and are less than adequate. It is therefore very important for reliable senescent cell biomarkers to be identified, otherwise it is impossible to confirm if an intervention is effective.
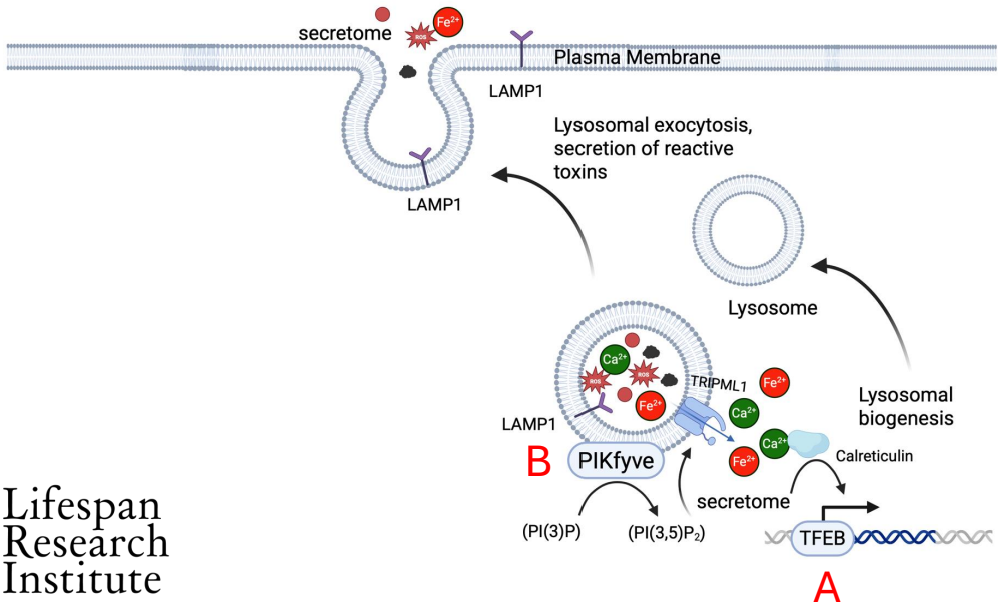
Senescent cells have increased lysosomal function. Lysosomes are bubble-shaped organelles that engulfs cellular waste and removes it from the cell. It does this by merging with the outer membrane to eject the waste through a process known as lysosomal exocytosis.
Researchers at LRI have used a database of membrane proteins to identify a protein called LAMP1. This protein is only present in healthy cells for a short time during lysosomal exocytosis, but in senescent cells, it persists.
Dr. Sharma and his team found a correlation between the expression of LAMP1 and senescence associated genes, including p21 and p16. They tested this association in human fibroblasts in a number of ways and found that LAMP1 expression increased significantly in senescent cells.
Untreated cells only expressed LAMP1 around 1% of the time, while cells that had induced senescence saw expression 20-60% of the time. How much depended on how senescence was induced in the cells.
We then investigated the possibility of utilizing LAMP1 as a therapeutic target. The team used an antibody-drug conjugate (ADC), which is comprised of an antibody aimed at a particular cell surface protein. The ADC was used to target LAMP1, which caused the senescent cells to die but didn’t affect healthy cells.
Ultimately, this opens up the possibility that LAMP1 could be a target for senolytic therapies that help clear harmful senescent cells. These are just two examples of how LRI is making important advances and driving progress in the aging research field.
Rejuvenation biotechnology has an advocacy problem
Advocacy is one of the most important things in our field, yet it is often the most overlooked thing. Often, the focus is understandably on the research side of things, but this comes at the cost of effectively engaging with policymakers, the medical world, and the public. While doing the actual research and achieving breakthroughs is critical, we also need to combine this with effective advocacy and communication strategies.
For years, we have watched the field develop, and one thing that has always struck us is how different groups in our community are often at odds with each other when it comes to advocacy. This has often led to completely opposing strategies, a disorganized community, and an unclear message. This is obviously not ideal.
Some groups suggest that public engagement isn’t important at all and that the focus should be on the science until there is something to show. Other groups believe that we should be engaging the public to pave the way for the emerging technologies that are to arrive. At face value, both of these seem like reasonable positions to take.
There may be a middle ground between both approaches, but how would we know, and how can we better understand public sentiment?
Developing a cultural intelligence platform for advocacy
This is why we are supporting the development of a cultural intelligence platform by the Public Longevity Group (PLG). Its goal is to create tools to gauge public sentiment, measure the impact of media coverage, study social media activity relating to the field, and develop optimal communication strategies to reach as-yet-untapped audiences.
In this video, Sho, the founder of PLG, highlights the importance of public trust in promoting rejuvenation biotechnology. He emphasizes that mere scientific advancements are insufficient; cultural acceptance and comprehension are vital. PLG employs data-driven techniques to assess public opinion, monitor discussions, and craft engaging narratives that appeal to various demographic groups.
By refining communication strategies and building partnerships with organizations like the Lifespan Research Institute, PLG aims to bridge the gap between science and society, ensuring that advancements are not only scientifically possible but are socially accepted.
Scientific narratives focusing on rejuvenation and longevity are gaining traction, while lifestyle-based messaging is plateauing, suggesting a shift in public interest towards more advanced interventions. Now is the perfect time to develop the tools to optimize our advocacy efforts.
Key points of the project:
The research is advancing, but for it to have a meaningful impact, gaining public trust, cultural acceptance, and legitimacy are essential. Without these, scientific breakthroughs may fail to reach the public or gain momentum.
Framing longevity in terms of long-term health and independence can significantly increase public support, shifting attitudes from opposition to acceptance.
The cultural intelligence platform will use strategic methods like tracking online discourse, conducting high-resolution surveys, and analyzing consumer behavior to understand public sentiment around longevity.
The cultural intelligence platform will also identify underserved regions and demographics, revealing new opportunities for targeted outreach.
Data-driven experiments will be used to test which narratives and communication strategies resonate across different demographics and political identities.
Finding new ways to communicate about the science using fresh metaphors, entry points, and language that engages the public without diluting the mission.
Recognizing the need for data-driven cultural insights to foster public engagement and collaboration within the rejuvenation and longevity field. This is a group effort, and we need to work together to make things happen.
PLG is already collaborating with key organizations like the Alliance for Longevity Initiatives and the University of Texas Medical Branch to turn cultural insights into actionable strategies for advancing longevity science.
The cultural intelligence platform campaign is live on the LRI website now; please help us to build the tools our movement needs to succeed! Thank you to everyone who has already stepped up to support this important initiative.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
Aging-related epigenetic changes result in decreased capacity to maintain cellular identity [2], and some studies suggest aging-related changes in cellular states [3].
Such changes to the cell state can be a target for rejuvenation strategies. Probably the most famous rejuvenation strategy is the use of the Yamanaka factors, OCT4, SOX2, KLF4, and c-MYC (OSKM), which can rejuvenate fully differentiated aged cells into pluripotent stem cells; however, there is much to be understood regarding the mechanisms driving this rejuvenation.
The authors of this study focus specifically on the process of mesenchymal-epithelial transition (MET), which initiates the rejuvenation of fibroblasts into a pluripotent state by OSKM.
Drifting into old age
The authors analyzed gene expression in human tissue biopsies to identify aging-related trends. They noted some trends that are common among tissues and already known to be associated with aging, such as the aging-related increase in inflammation.
One specific observation, common across the tissues, captured the researchers’ attention: upregulation of pathways related to epithelial-mesenchymal transition (EMT). Given the common nature of this process across aging tissues, they named it ‘‘mesenchymal drift’’ (MD). They defined this as a process during which “cells undergo a partial or complete transition, leading to a compromised original cellular identity while acquiring new or intensifying existing mesenchymal traits, including altered extracellular matrix (ECM) production, cellular junction, tissue mechanical stiffening, and cytokine production.” They suggest that mesenchymal drift contributes to age-related organ dysfunction and might contribute to age-related diseases.
To investigate the latter, they analyzed available data from human biopsies obtained in various studies. They observed an increased expression, to a varying degree, of mesenchymal drift-associated genes in the disease-affected tissues taken from patients with lung diseases, liver diseases, chronic kidney diseases, cardiomyopathies, Alzheimer’s disease, osteoarthritis, and inflammatory bowel diseases as well as aged skin and endothelial cells from calcified atherosclerotic cores.
Prognostic marker
Because of the observed associations between multiple aging-associated diseases and mesenchymal drift, the researchers investigated its role as a predictor of clinical outcomes. They focused on idiopathic pulmonary fibrosis (IPF) in order to establish a proof of concept.
They divided IPF patients into low, middle, and high mesenchymal drift gene expression groups. The median survival of those groups was inversely correlated with the magnitude of mesenchymal drift. This suggests that mesenchymal drift genes can be used as prognostic markers and therapeutic targets.
Rejuvenating potential
Since the data obtained so far only establishes associations between the two observations, the researchers investigated the causality between mesenchymal drift and aging. They asked if biological age, as measured by epigenetic clocks, would be changed if they modulate the regulator of mesenchymal drift.
They re-analyzed publicly available data from breast cancer cell experiments that repressed ZEB1, a key EMT transcription factor that suppresses epithelial markers. Two epigenetic clocks (Horvath’s and PhenoAge) showed that repressing ZEB1 resulted in reduced biological age, suggesting that targeting mesenchymal drift has rejuvenating potential.
With this in mind, the researchers analyzed plasma proteins for potential drivers of mesenchymal drift across organs. They used UK Biobank data to identify mortality-associated plasma proteins. that have a stronger association with mortality were enriched in EMT pathways. They suggest that these findings show a significant association between mesenchymal drift regulators, aging, and increased mortality and that these plasma proteins are possible factors in driving mesenchymal drift across organs.
Rejuvenating without pluripotency
These researchers also aimed to mitigate mesenchymal drift using the Yamanaka factors, which are famous for their rejuvenating effects. Previous research has found that differentiated cells dedifferentiate by going through MET during the Yamanaka factor-driven rejuvenation process. [4] To learn more about the transition between the mesenchymal and epithelial states in aged cells, the researchers measured gene expression in human fibroblasts from a 96-year-old donor at different time points following OSKM induction.
Among different paths taken by cells, they identified a “partially reprogrammed” trajectory that shows some rejuvenation features. This trajectory was characterized by downregulation of mesenchymal genes in the first days following OSKM induction, while reprogramming-associated epithelial genes “were only beginning to be upregulated toward the pluripotency state.” Such a response suggests a critical window during early partial reprogramming that plays an essential role in the dampening of mesenchymal drift through partial MET.
Experiments in cell cultures with a non-functional OCT4 ene, which are unable to be reprogrammed to pluripotent stem cells, showed that those cells are still able to suppress the mesenchymal drift gene program following the expression of the three other Yamanaka factors. Those results suggest that mitigating mesenchymal drift is a potential mechanism behind rejuvenation, independent of inducting pluripotency.
Partial reprogramming’s impact on tissues and cells
Promising results obtained in the cell cultures led to further research in mice. First, in naturally aged mice, long-term (over 7 months) OSKM induction led to significantly decreased mesenchymal drift gene expression in the kidney and liver, and the trend toward decrease was also observed in most other organs. Short-term (around 1 month) OSKM induction at an advanced age significantly reduced mesenchymal drift in the spleen and showed a trend towards a decrease in the liver. Similar results (downregulation of mesenchymal drift in the bone marrow compartment and a similar trend in the skin and spleen) were seen in progeroid mice treated with a virus expressing OSK for two and a half months.
A cellular analysis followed the tissue-level analysis. The researchers used the available data from different studies regarding the mouse intestine and pancreas gene expression following a few days of OSKM induction. In the intestine, most cell types showed mesenchymal drift gene expression reduction, which the researchers linked to improved intestinal regenerative capacity. However, in the pancreas, they observed increased mesenchymal drift gene expression in most cell types; however, some subpopulations showed a decrease, suggesting cell-type-specific responses to OSKM induction that call for optimization of the Yamanaka factors induction duration and cell-type-specific targeting for optimal effects in further therapies.
The researchers concluded that their results show “that the partially reprogrammed state exhibited a notable decrease in the expression of mesenchymal drift genes through partial MET.” While those results are promising, this research was conducted on cell cultures and animal models and needs further validation in human trials.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
[1] Lu, J. Y., Tu, W. B., Li, R., Weng, M., Sanketi, B. D., Yuan, B., Reddy, P., Rodriguez Esteban, C., & Izpisua Belmonte, J. C. (2025). Prevalent mesenchymal drift in aging and disease is reversed by partial reprogramming. Cell, S0092-8674(25)00853-0. Advance online publication.
[2] Hernando-Herraez, I., Evano, B., Stubbs, T., Commere, P. H., Jan Bonder, M., Clark, S., Andrews, S., Tajbakhsh, S., & Reik, W. (2019). Ageing affects DNA methylation drift and transcriptional cell-to-cell variability in mouse muscle stem cells. Nature communications, 10(1), 4361.
[3] Izgi, H., Han, D., Isildak, U., Huang, S., Kocabiyik, E., Khaitovich, P., Somel, M., & Dönertaş, H. M. (2022). Inter-tissue convergence of gene expression during ageing suggests age-related loss of tissue and cellular identity. eLife, 11, e68048.
[4] Waryah, C., Cursons, J., Foroutan, M., Pflueger, C., Wang, E., Molania, R., Woodward, E., Sorolla, A., Wallis, C., Moses, C., Glas, I., Magalhães, L., Thompson, E. W., Fearnley, L. G., Chaffer, C. L., Davis, M., Papenfuss, A. T., Redfern, A., Lister, R., Esteller, M., … Blancafort, P. (2023). Synthetic Epigenetic Reprogramming of Mesenchymal to Epithelial States Using the CRISPR/dCas9 Platform in Triple Negative Breast Cancer. Advanced science (Weinheim, Baden-Wurttemberg, Germany), 10(22), e2301802.
Date Posted: September 17, 2025
Comments Off on Lifespan Research Institute Launches Public Longevity Group
[Mountain View, September 17, 2025] — Lifespan Research Institute (LRI) today announced the launch of the Public Longevity Group (PLG), a new initiative focused on bridging the cultural gap between scientific breakthroughs in aging and their public acceptance. To kickstart its work, PLG has opened a crowdfunding campaign to develop tools that measure and strengthen public trust in longevity science.
While the science of longevity biotechnology continues to advance, skepticism and cultural resistance limit progress, with some studies showing that more than half of Americans would reject a safe, proven therapy to extend life. This hesitation poses risks of raising costs, delaying health-promoting regulation, and slowing the delivery of treatments that could combat age-related diseases and extend healthy lifespan.
“The breakthrough that unlocks all other breakthroughs is public trust,” said Sho Joseph Ozaki Tan, Founder of PLG. “Without it, even the most promising therapies may never reach the people they’re meant to help. PLG exists to change that.”
“Persuasion is a science too,” said Keith Comito, CEO of Lifespan Research Institute. “To bring health-extending technologies to the public as quickly as possible, we must approach advocacy with the same rigor as our research. With PLG, we’ll be able to systematically measure and increase social receptivity, making the public’s appetite for credible longevity therapies unmistakable to policymakers, investors, and the public itself.”
PLG is developing the first data-driven cultural intelligence system for longevity—a platform designed to track real-time sentiment, test narratives, and identify which messages resonate and which backfire. Early tools include:
The Longevity Cultural Clock: a cultural barometer mapping readiness and resistance across demographics and regions.
Sentiment Dashboards: real-time monitoring of public, investor, and policymaker perceptions.
Narrative Testing Tools: data-driven analysis that will enable robust pathways to public support.
The crowdfunding campaign will provide the initial $100,000 needed to launch these tools, creating the cultural foundation required for healthier, longer lives.
With a lean, data-driven team, the group aims to provide open-access cultural insights for advocates and policymakers while offering advanced analytics to mission-aligned partners.
The PLG campaign is sponsored by the members of LRI’s Lifespan Alliance, a consortium of mission-aligned organizations that believe in the promise of extending healthy human lifespan. Newly-joined members include OpenCures, AgelessRx, and Lento Bio.
About Lifespan Research Institute
Lifespan Research Institute accelerates the science and systems needed for longer, healthier lives by uniting researchers, investors, and the public to drive lasting impact. LRI advances breakthrough science, builds high-impact ecosystems, and connects the global longevity community.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
Scientists have demonstrated that even two days on a Western-like high-fat diet reduce hippocampal glucose availability, which activates a subset of inhibitory neurons and causes memory problems in mice [1].
Is it OK to eat junk occasionally?
Metabolic disease and obesity are known to harm cognitive function and have been linked to neurodegenerative diseases such as Alzheimer’s [2], among numerous other health problems. However, many people tend to think that they can get away with temporarily switching to an unhealthy diet or even with constantly eating such a diet while maintaining a healthy weight.
Recent research shows that it’s not that simple: even short periods of a high-fat diet can cause issues, including a decline in cognitive function [3]. A new study from the University of North Carolina, published in Neuron, delves deeper into this phenomenon, yielding intriguing insights.
Memory impairments after just two days
The researchers set out to investigate the impact on healthy adult mice of a very short-term high-fat diet (stHFD) consisting of 58% fat, 25% carbohydrates, and 17% protein. According to the paper, this diet resembles Western diets rich in saturated fat.
Two days into the experiment, the researchers put the mice through a battery of cognitive tests and found that mice on this stHFD did noticeably worse on some of them, particularly on novel place recognition (NPR) and contextual fear conditioning (CFC), indicating “significant impairments in hippocampus-dependent spatial and contextual memory.” Importantly, body weight and blood glucose levels remained unchanged due to the short duration of the intervention.
The stHFD mice, however, were more physically active in the arena, traveling farther (and/or at higher speed) during the open-field session. This suggests that the poorer scores on the two cognitive tests were not due to the mice being lethargic or motor-impaired.
The neurons that didn’t get enough glucose
NPR is thought to depend on the function of the hippocampal region called the dentate gyrus, and the hippocampus is especially vulnerable to brief HFD. The team detected greatly increased activity of a subset of neurons called cholecystokinin-expressing interneurons (CCK-INs) in the dentate gyrus. Interneurons are local inhibitory neurons. Stimulating CCK-INs impaired memory in controls, while dampening their signals rescued memory deficits in stHFD mice.
Prior research has found that just three days of HFD downregulates GLUT1, the main endothelial glucose transporter, across the brain, including the hippocampus. With less GLUT1 at the blood-brain barrier, less glucose gets from capillaries into brain tissue, as if a faucet is turned down, so neuronal glucose availability drops even if blood glucose is normal.
The researchers confirmed that CCK-INs in the dentate gyrus sit farther from capillaries than granule neurons and hence have worse access to the reduced amount of glucose. CCK-INs are glucose-inhibited, which means that when glucose is low, their activity increases, inhibiting that of other local neurons. Giving stHFD mice extra glucose or putting them on a one-night fasting-refeeding protocol restored the activity of CCK-INs. Crucially, if the researchers forced CCK-INs to stay active by chemogenetic activation, the glucose/refeeding no longer improved memory.
PKM2 inhibition rescues memory
Investigating what intracellular switch links low glucose to CCK-IN hyperactivity, the team focused on a glycolytic enzyme, PKM2. stHFD elevated PKM2 signaling markers in CCK-INs, while knocking down PKM2 in these cells reduced their baseline activity and rescued NPR without changing intake/weight. Shikonin, a PKM2 inhibitor, also rescued NPR.
“We knew that diet and metabolism could affect brain health, but we didn’t expect to find such a specific and vulnerable group of brain cells, CCK interneurons in the hippocampus, that were directly disrupted by short-term high-fat diet exposure,” said Juan Song, PhD, principal investigator, professor of pharmacology at the UNC School of Medicine, and a senior author of the study. “What surprised us most was how quickly these cells changed their activity in response to reduced glucose availability and how this shift alone was enough to impair memory.”
Having discovered this pathway for diet-related memory deficiency, the team asked whether this can be applied beyond stHFD. In a “classic” scenario of obesity induced by 10 weeks of HFD, mice showed spatial-memory deficits, but chronic CCK-IN inhibition for the duration of HFD or long-term PKM2 knockdown reversed these effects.
“This work highlights how what we eat can rapidly affect brain health and how early interventions, whether through fasting or medicine, could protect memory and lower the risk of long-term cognitive problems linked to obesity and metabolic disorders,” said Song. “In the long run, such strategies could help reduce the growing burden of dementia and Alzheimer’s linked to metabolic disorders, offering more holistic care that addresses both body and brain.”
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
[1] Landry, T., Perrault, L., Melville, D., Chen, Z., Li, Y. D., Dong, P., … & Song, J. (2025). Targeting glucose-inhibited hippocampal CCK interneurons prevents cognitive impairment in diet-induced obesity. Neuron.
[2] Patel, V., & Edison, P. (2024). Cardiometabolic risk factors and neurodegeneration: a review of the mechanisms underlying diabetes, obesity and hypertension in Alzheimer’s disease. Journal of Neurology, Neurosurgery & Psychiatry, 95(6), 581-589.
[3] McLean, F. H., Grant, C., Morris, A. C., Horgan, G. W., Polanski, A. J., Allan, K., … & Williams, L. M. (2018). Rapid and reversible impairment of episodic memory by a high-fat diet in mice. Scientific reports, 8(1), 11976.
Date Posted: September 15, 2025
Comments Off on How Macrophages Manage Obesity and Change With Age
Macrophages declining with age is a well-known problem, and many proposed solutions involve changing their polarity from M1, which favors inflammation, to M2, which favors regeneration. However, this does not apply to adipose tissue macrophages (ATMs), which do not polarize themselves in this way [1]. These tissue-macrophages have subsets, including lipid-associated macrophages (LAMs) [2] and vasculature-associated macrophages (VAMs) [3]. These researchers note that little work has been done on relating these various subtypes of ATMs to aging.
This paper addresses several of these subtypes, including nerve-associated macrophages (NAMs), which not only police the nerves themselves [4] but are linked to the functions of the tissues where the nerves are located, such as the gut [5]. In the fat tissue, NAM behavior has been linked to obesity [6].
Macrophage subtypes can be clustered
The experiments began by deriving visceral white adipose tissue (VAT) from mice and isolating immune cells that were positive for both F4/80 and CD11b+. The mice were either 2 months or 22 months of age, and both male and female mice were used in this experiment.
About three-quarters of these cells were found to be tissue resident, with the other quarter flowing in the vasculature. Principal component analysis found that there were substantial differences between circulating and resident cells, and there were also found to be differences between the cells found in brown fat and white fat.
The researchers then clustered these cells based on single-cell RNA sequencing. This method allowed them to find and discard contaminating cells as well as identify cellular commonalities and differentiation into subtypes. Interestingly, the most numerous cluster was sharply different between sexes; a cluster that was very common in male mice, Cluster 0, was sharply reduced in females, and an exceptionally common female cluster, Cluster 5, was much less common in males.
Cluster 5 was found to be highly enriched for Maoa, which breaks down catecholamines such as neurotransmitters. This and other evidence led the researchers to conclude that these are NAMs. A similar gene expression analysis revealed that Cluster 0 is composed of VAMs.
Age changes the landscape
These clusters were significantly changed with aging, according to a flow cytometry analysis focusing on surface markers, and how they did so was largely dependent on sex. Between the 2-month-old and 22-month-old mice, Cluster 0 decreased with aging in males but not females, Cluster 5 decreased in females but not males, and cluster 13 increased in females but not males. Clusters 4 and 10 increased in both sexes, and Cluster 4 was recognized solely as being associated with aging rather than tissue function; the researchers called these aging-associated macrophages (AAMs), which express pro-inflammatory compounds and are characterized by the age-related marker CD38.
Interestingly, these flow cytometry results did not entirely agree with the RNA sequencing analysis, which suggested that the number of Cluster 5 NAMs remained unchanged in female mice. It is plausible that cell surface marker expression changed with aging.
NAMs were found to be almost uniquely associated with aging. While the AAMs in Cluster 4 were also found to have senescent-like features, these features were common in Cluster 5 NAMs. This was based on gene expression analysis and was relative to the behavior of aged cells rather than directly related to the known cellular markers of senescence.
A link to obesity
However, as the researchers discovered, these cells perform critical tasks. Specifically, adipose-derived NAMs were found to help maintain myelin, the cellular sheaths that protect neuronal axons. Depleting the adipose-derived NAMs of 3-month-old mice led to dysregulation of catecholamines as well as obesity; freely fed mice that had adipose-derived NAMs depleted were considerably more likely to gain weight. Further experimentation with older mice found that NAM depletion led to significant increases in inflammation.
This research highlights a small part of the heterogeneity found in biology, details its changes with aging, and suggests that it may be possible to specifically handle macrophage subtypes; for example, future experiments may find that it is beneficial to reduce the population of aging-associated macrophages as identified here. This focus on macrophages may also be valuable in examining the fundamental causes of obesity and analyzing the effects of existing anti-obesity drugs.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
[1] Camell, C. D., Sander, J., Spadaro, O., Lee, A., Nguyen, K. Y., Wing, A., … & Dixit, V. D. (2017). Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature, 550(7674), 119-123.
[2] Jaitin, D. A., Adlung, L., Thaiss, C. A., Weiner, A., Li, B., Descamps, H., … & Amit, I. (2019). Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell, 178(3), 686-698.
[3] Silva, H. M., Báfica, A., Rodrigues-Luiz, G. F., Chi, J., Santos, P. D. E. A., Reis, B. S., … & Lafaille, J. J. (2019). Vasculature-associated fat macrophages readily adapt to inflammatory and metabolic challenges. Journal of Experimental Medicine, 216(4), 786-806.
[4] Kolter, J., Feuerstein, R., Zeis, P., Hagemeyer, N., Paterson, N., d’Errico, P., … & Henneke, P. (2019). A subset of skin macrophages contributes to the surveillance and regeneration of local nerves. Immunity, 50(6), 1482-1497.
[5] Muller, P. A., Koscsó, B., Rajani, G. M., Stevanovic, K., Berres, M. L., Hashimoto, D., … & Bogunovic, M. (2014). Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell, 158(2), 300-313.
[6] Pirzgalska, R. M., Seixas, E., Seidman, J. S., Link, V. M., Sánchez, N. M., Mahú, I., … & Domingos, A. I. (2017). Sympathetic neuron–associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nature medicine, 23(11), 1309-1318.
Date Posted: September 12, 2025
Comments Off on Microplastics Cause Cognitive Deficits in APOE4 Mice
Exposure to tiny particles that plastic products shed (microplastics) has been linked to increased mortality and diseases [2]. Microplastics are ubiquitous and enter organs, including the brain, triggering inflammation. A recent study showed alarming levels of microplastic accumulation in human brains [3]. However, rigorous studies of the exact effects of microplastics on the brain have been scarce.
A new study by researchers at the University of Rhode Island College of Pharmacy asked a question whether microplastics exposure can promote Alzheimer’s disease in mice genetically predisposed to it. The team created genetically modified mice that were homozygous for either the human APOE ε3 or APOE ε4 allele. The latter is strongly associated with an increased risk of Alzheimer’s, while the former is considered “normal,” neither increasing risk nor providing protection. Both sexes were included to avoid missing sex-specific effects.
At 3-6 months of age, before developing pathologies, mice were randomized into control or exposure groups, with 8 animals per each combination of sex, genotype, and condition. Then, for three weeks, the treatment groups drank water containing a mix of fluorescent polystyrene particles at two sizes: 0.1 μm (nanoplastic) and 2 μm (microplastic), at 0.125 mg/mL. The chosen dose was intentionally high relative to what the average human receives from the environment. This was done to compensate for the short duration of exposure.
“In these mice, like in people, it’s not a guarantee that you’re going to see any changes in cognition. You could have identical twins both carrying APOE4, one totally cognitively healthy, and the other could develop Alzheimer’s disease,” said URI pharmacy assistant professor Jaime Ross, the lead author of the study.
Males more apathetic, females more forgetful
The team then ran a battery of cognitive tests. In the open field test, cognitively normal mice are expected to spend little time in the open, which is these rodents’ natural behavior helping them to avoid predators. Male APOE ε4 mice who were exposed to microplastics, however, spent much more time in the center of the arena, a pattern that the authors interpret as apathy-like cognitive disruption.
Females did not show this pattern. However, in a different test, novel object recognition (NOR), it was the female APOE ε4 mice who showed poorer recognition memory. Several other tests produced null results, but the authors suggest it actually shows that, just like in human Alzheimer’s, the effect was less “broad anxiety” and more cognition-focused.
Ross tied the findings to known sex-related differences in Alzheimer’s symptoms in humans. “In human Alzheimer’s patients,” she said, “men tend to experience more changes in apathy; they care less. Women experience more changes in memory. So, the memory and the apathy connection are pretty clear: when you expose animals that are carrying the largest known risk factor in humans for developing Alzheimer’s disease to micro- and nanoplastics, lo and behold, their behavior changes in a sex-dependent manner similar to the sex-dependent differences we see with Alzheimer’s patients.”
“Similar to what we’re seeing in the real world”
The plastic particles were confirmed to reach the brain, at least the larger ones (0.1 μm particles are below histology detection limits), and the researchers attempted to analyze possible mechanisms behind the impact on cognition. The team examined GFAP, a marker of astrocyte activation/health, and IBA1, a microglial marker, but the results were inconclusive. The researchers suggest that microplastics exert their effect on cognition not via classical microglial inflammation and call for further research.
Two important caveats apply. First, the high dose and short exposure limits the study’s generalizability. Second, real-world plastics are weathered, chemically varied, and often carry adsorbed pollutants. This study used pristine polystyrene spheres, which do not mirror these conditions.
“So, that tells us there’s something about lifestyle, something about the environment going on,” Ross said. “There are modifiable factors we’re studying related to Alzheimer’s – diet, exercise, vitamins, and especially environmental toxins like microplastics. If you carry the APOE4, and you happen to consume a lot of microplastics, will this contribute to Alzheimer’s disease?”
“There has not been a lot of money spent on the human health impacts of microplastics,” she added. “It’s interesting that what we’re seeing in mice is similar to what we’re seeing in the real world. We want to encourage further research into the scourge of micro- and nanoplastics.”
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
[1] Gaspar, L., Bartman, S., Tobias-Wallingford, H., Coppotelli, G., & Ross, J. M. (2025). Short-term exposure to polystyrene microplastics alters cognition, immune, and metabolic markers in an apolipoprotein E (APOE) genotype and sex-dependent manner. Environmental Research Communications, 7(8), 085012.
[2] Marfella, R., Prattichizzo, F., Sardu, C., Fulgenzi, G., Graciotti, L., Spadoni, T., … & Paolisso, G. (2024). Microplastics and nanoplastics in atheromas and cardiovascular events. New England Journal of Medicine, 390(10), 900-910.
[3] Nihart, A. J., Garcia, M. A., El Hayek, E., Liu, R., Olewine, M., Kingston, J. D., … & Campen, M. J. (2025). Bioaccumulation of microplastics in decedent human brains. Nature medicine, 31(4), 1114-1119.
Date Posted: September 12, 2025
Comments Off on LongX Hosts the Youth in Longevity Biotech Showcase
On September 18, 2025, Longevity Xplorer (LongX) will be hosting the first-ever “Youth in Longevity Biotech Showcase”, a virtual event featuring lightning talks from young professionals in longevity fellowships around the world. The event will cover fellow contributions and highlight the importance in supporting the next generation of leaders in aging biology. LongX welcomes the entire longevity community to attend.
A remote fellowship designed to help early career professionals apply themselves in the longevity biotech sector through courses, mentorship, and industry placements.
A program for undergraduate students and recent graduates to conduct biomedical research on age-related diseases under the guidance of a scientific mentor, with an emphasis on developing both laboratory and communication skills.
A 1-2 year program for recent college graduates to gain intensive research experience in aging and age-related diseases at the Buck Institute before pursuing an advanced degree or a career in the biotech industry.
An aging biology fellowship that serves as a talent accelerator, supporting ambitious future leaders with knowledge, community, mentorship, and project support.
This event serves to:
Promote cross-pollination between aging biology-focused fellowships
Improve transparency in program developments and outcomes
Increase support for aging biology training programs
Foster a networking space for those interested in talent and education in longevity
About LongX
LongX was launched in 2023 as a platform for youth interested in longevity. We prioritize fostering innovation and interdisciplinary collaboration, aiming for both short-term impact and long-term progress. We encourage exploration beyond traditional roles and aim to equip future experts with the skills to drive progress. Our Substack provides regular articles on our thoughts, experiences, and interviews.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
These researchers begin by discussing epigenetic clocks and how much the measured alterations in gene expression cause other aspects of aging, the details of which remain unclear [1]. Accelerated aging, as measured by these clocks, is associated with cardiometabolic diseases [2]; if altering the expression of clock-related genes is demonstrated to have benefits in this area, it could pave the way for interventions; if this method is demonstrated not to be beneficial, then they may remain useful simply as clocks. There is also the question of generalizability: whether or not clocks are equally valuable across populations.
Limited connections between clocks and metabolites
Here, the researchers focused on obese people by using data from the MACRO trial, which was conducted to test the effects of weight loss interventions between 2008 and 2011 [3].
This trial utilized 148 participants between 22 and 75 years of age who had high BMI indices (between 30 and 45) but did not have such metabolic diseases as cardiovascular disease or type 2 diabetes. They were divided into low-carbohydrate and low-fat groups; the low-carb group was reported to have more weight loss than the low-fat group. Adherence to these diets was high.
These researchers used three samples from each person: at baseline, 3 months, and 12 months. To minimize noise, two principal component-based clocks, PCPhenoAge and PCGrimAge, were used along with the DunedinPACE clock that natively measures age acceleration. Age, sex, ethnicity, body weight, education, smoking, and alcohol use were all factored in as covariates.
Interestingly, lower total cholesterol was found to be associated with more rapid aging, according to both PCPhenoAge and DunedinPACE. Other metabolic factors were found to be associated with accelerated aging, including reduced levels of ghrelin and adiponectin along with higher levels of insulin, the insulin resistance marker HOMA-IR, and C-reactive protein (CRP). PCGrimAge did not have any statistically significant association between metabolic factors and epigenetic aging.
After dietary interventions, however, most of these correlations disappeared; the only ones that remained were the low adiponectin and the high CRP according to the DunedinPACE clock.
The interventions themselves had noticeable effects. Only three months of dieting did not yield any statistically significant effect on epigenetic aging. However, after 12 months, despite the dets being more effective for weight loss, PCPhenoAge and PCGrimAge reported increased epigenetic aging in the low-carb group compared to the low-fat group. DunedinPACE, however, reported that epigenetic age acceleration was decreased in both groups.
As expected, the dietary interventions had significant benefits for metabolic issues. However, critical to this study, the reseachers found that none of these benefits were mediated by epigenetic alterations as measured by these clocks: “Specifically, all indirect effects and proportions mediated were non-significant (FDR > 0.05), suggesting that the observed associations were not driven through changes in epigenetic aging.”
A clock may not always reflect health
These results, or lack thereof, led the researchers to suggest that the health benefits of moderately rapid weight loss are not well-captured in epigenetic clocks designed to measure aging. Specifically, they note that there is an “uncoupling” between the metabolic benefits observed in these studies and clock measurements. They further suggest that this may be due to the time periods involved; DunedinPACE “may be more indicative of cumulative biological burden that changes gradually over time” and less sensitive to relatively short-term effects.
The researchers highlight that their study is in line with previous work on this topic; the CALERIE trial found that caloric restriction, a well-known longevity intervention, had some effects on DunedinPACE but fewer effects on PCPhenoAge and PCGrimAge [4]. In total, their findings led the researchers to specifically warn against the use of epigenetic clocks as surrogate endpoints in metabolism-related interventions, suggesting instead that they “should remain exploratory and be interpreted with more established physiological markers.”
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.
[1] Ferrucci, L., Barzilai, N., Belsky, D. W., & Gladyshev, V. N. (2025). How to measure biological aging in humans. Nature Medicine, 1-1.
[2] Lo, Y. H., & Lin, W. Y. (2022). Cardiovascular health and four epigenetic clocks. Clinical Epigenetics, 14(1), 73.
[3] Bazzano, L. A., Hu, T., Reynolds, K., Yao, L., Bunol, C., Liu, Y., … & He, J. (2014). Effects of low-carbohydrate and low-fat diets: a randomized trial. Annals of internal medicine, 161(5), 309-318.
[4] Waziry, R., Ryan, C. P., Corcoran, D. L., Huffman, K. M., Kobor, M. S., Kothari, M., … & Belsky, D. W. (2023). Effect of long-term caloric restriction on DNA methylation measures of biological aging in healthy adults from the CALERIE trial. Nature Aging, 3(3), 248-257.
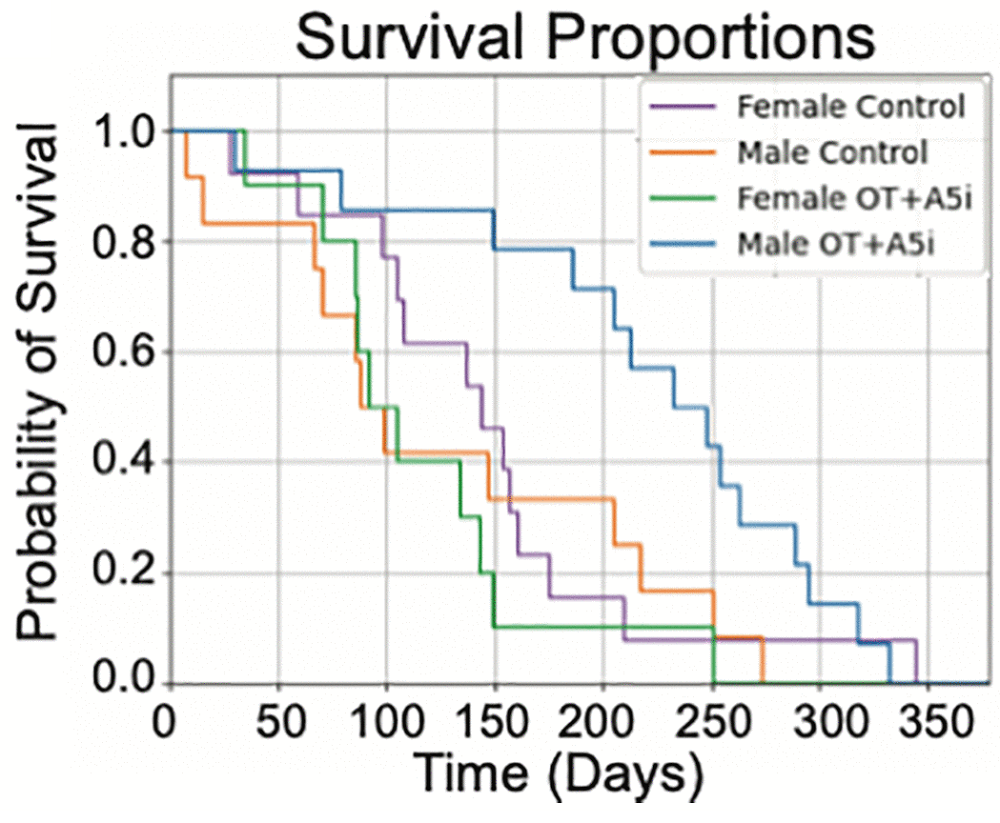

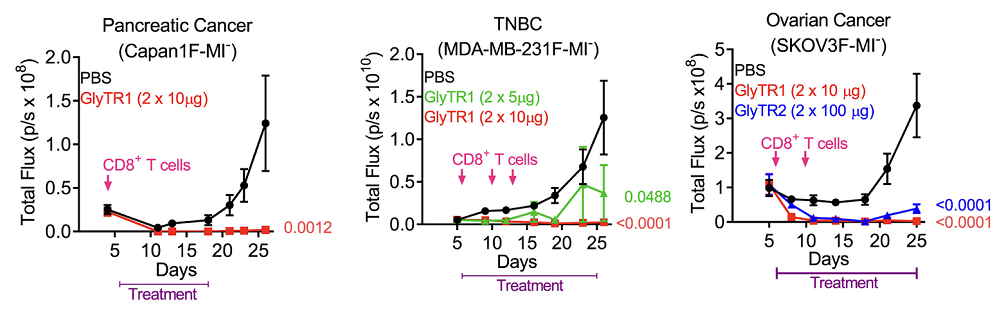
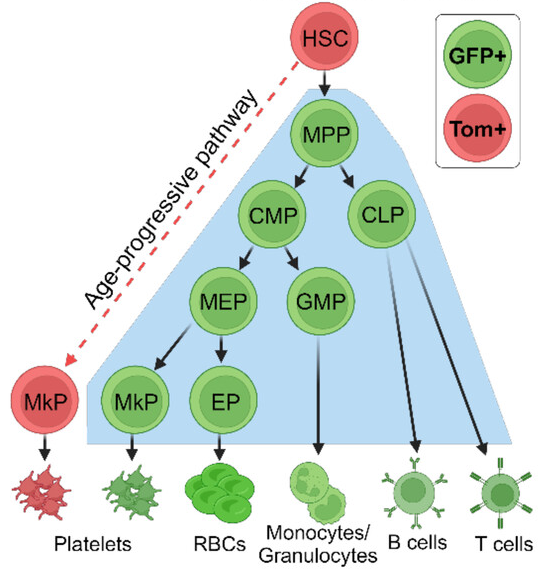


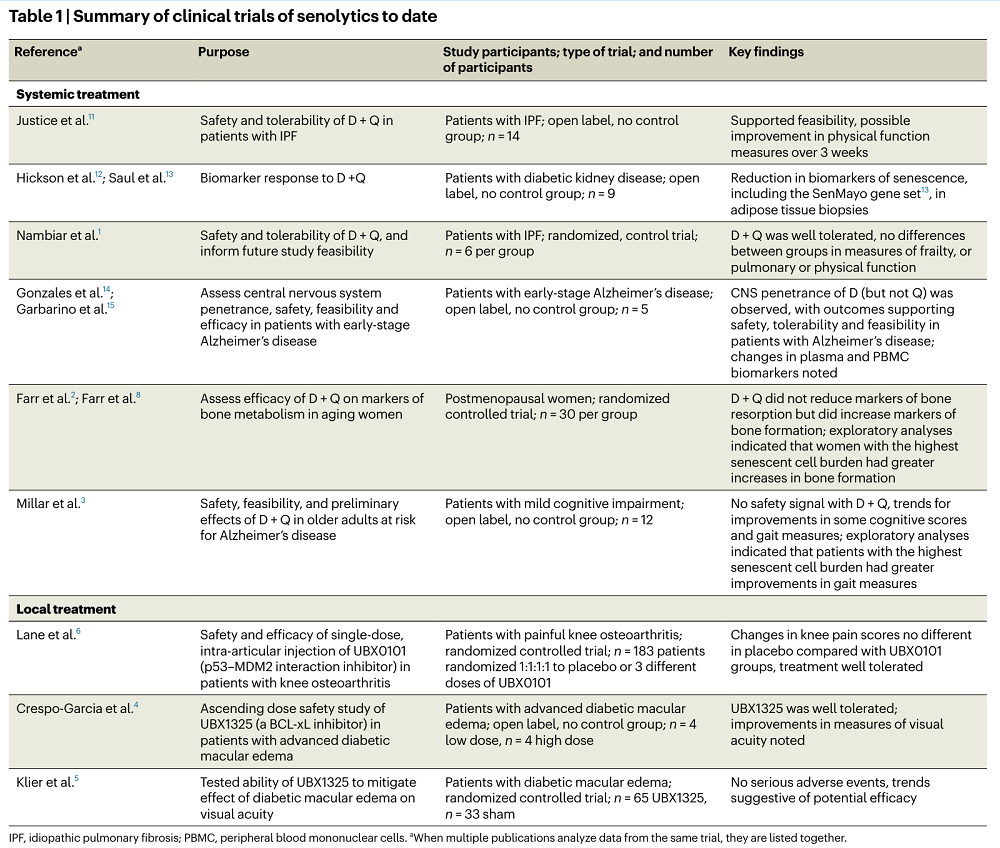
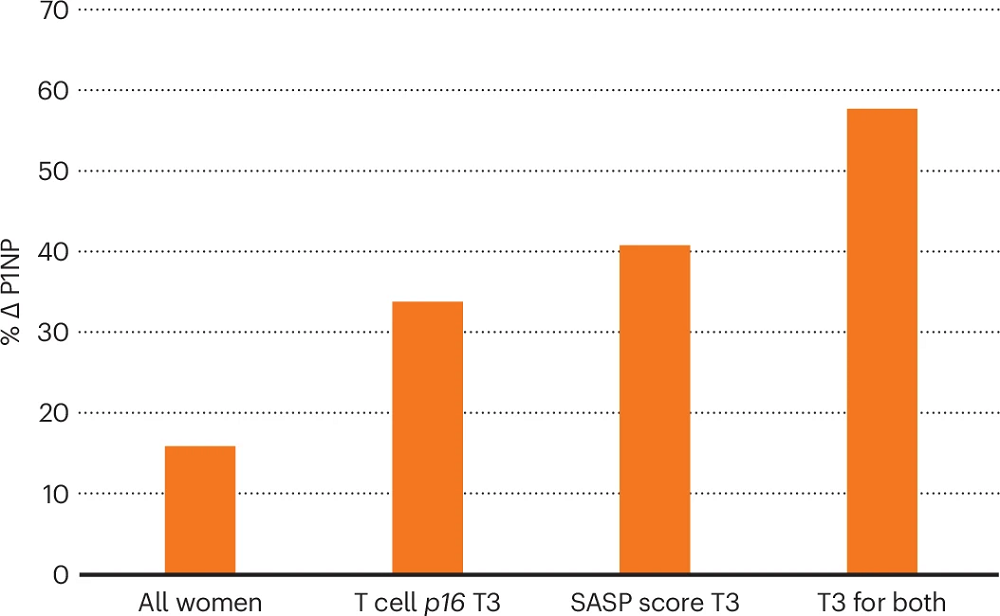
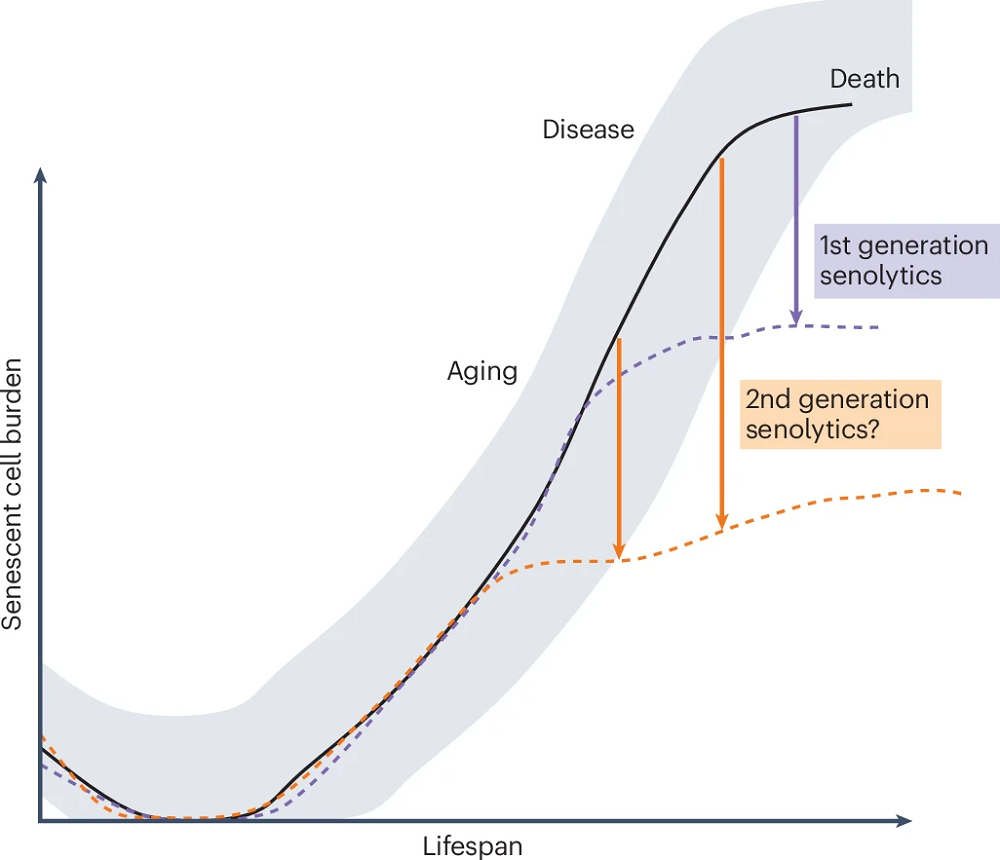




 Two people nearly died, and several more sought treatment, after receiving peptide injections at the last RAADfest in Las Vegas. This highlighted some well-known problems and contradictions in the longevity field.
Two people nearly died, and several more sought treatment, after receiving peptide injections at the last RAADfest in Las Vegas. This highlighted some well-known problems and contradictions in the longevity field. In the heart of San Francisco, a tower in the downtown area is transforming into a center for longevity, artificial intelligence, cryptocurrency, and robotics.
In the heart of San Francisco, a tower in the downtown area is transforming into a center for longevity, artificial intelligence, cryptocurrency, and robotics. Rejuve.AI is a company founded by Jasmine Smith and well known AI researcher Ben Goertzel. Recently, they have launched a new longevity app, and Arkadi caught up with them to find out more.
Rejuve.AI is a company founded by Jasmine Smith and well known AI researcher Ben Goertzel. Recently, they have launched a new longevity app, and Arkadi caught up with them to find out more. The Longevity Summit Dublin has become quite the go-to event in the longevity community. The event first launched in 2022 and has grown in popularity since then.
The Longevity Summit Dublin has become quite the go-to event in the longevity community. The event first launched in 2022 and has grown in popularity since then.