Researchers Rejuvenate the Thymi of Old Mice
Researchers have successfully demonstrated that rejuvenation of the aged thymus to make it functionally younger again is possible.
The thymus wastes away as we age
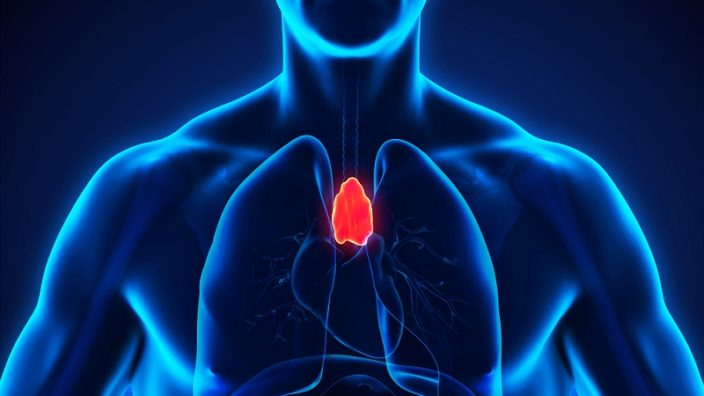
The thymus is a small organ located in the upper part of the chest directly behind the sternum and between the lungs. Before we are even born and through childhood, the thymus is busy producing and maturing T cells, immune cells that patrol the body and destroy invading pathogens. The thymus secretes a hormone known as thymosin, which is needed for the creation and maturation of T cells.
The thymus reaches its maximum weight of around one ounce during puberty but then starts to steadily shrink, and the immune-producing cells are replaced by fat cells; this process is known as thymic involution. By the time we reach old age, the thymus has all but turned to fat and ceased to produce immune cells. From then on, you are coasting along using the pool of T cells you have left, which the thymus created when you were younger.
One way researchers believe that the diseases of old age might be kept at bay is by rejuvenation of the thymus: in other words, reversing thymic involution so that the organ can continue to produce fresh T cells beyond puberty, into early adulthood, and potentially beyond. The decline of the immune system known as immunosenescence plays a key role in aging, so successful rejuvenation of the thymus could be a real game changer for continued health in old age.
A number of animal studies have already demonstrated the plausibility of rejuvenating the aged thymus, and there has even been a small human trial led by Dr. Greg Fahy in 2019, which successfully demonstrated that thymic involution can be reversed using a combination of growth hormone, metformin, and DHEA.
This first demonstration of age reversal in the human thymus was a huge step forward for the field. However, this may not be the most efficient way to achieve thymus rejuvenation, and the race is now on to find the optional way and translate that to humans.
The researchers of this new study created two kinds of FOXN1-reprogrammed embryonic fibroblasts and engrafted them directly into the thymi of aged mice. These young cells encouraged significant regrowth of these old thymi, which rejuvenated both organ structure and function.
The researchers also noted an increased level of thymopoiesis, the process that turns thymocytes into mature T cells. There was also a reduction of senescent T cells and auto-reactive T cell-mediated inflammation.
Age-associated systemic, chronic inflammation is partially attributed to increased self (auto)-reactivity, resulting from disruption of central tolerance in the aged, involuted thymus. This involution causally results from gradually decreased expression of the transcription factor FOXN1 in thymic epithelial cells (TECs), while exogenous FOXN1 in TECs can partially rescue age-related thymic involution. Given the findings that TECs induced from FOXN1-overexpressing embryonic fibroblasts can generate an ectopic de novo thymus under the kidney capsule and intra-thymically injected naturally young TECs can lead to middle-aged thymus regrowth, we attempted to extend these two findings by combining them as a novel thymic rejuvenation strategy with two types of promoter-driven (Rosa26CreERT and FoxN1Cre) Cre-mediated FOXN1-reprogrammed embryonic fibroblasts (FREFs). We engrafted these two-types of FREFs directly into the aged murine thymus. We found significant regrowth of the native aged thymus with rejuvenated architecture and function in both males and females, exhibiting increased thymopoiesis and reinforced thymocyte negative selection, along with reduced senescent T cells and auto-reactive T cell-mediated inflammation in old mice. Therefore, this strategy has preclinical significance and presents a strategy to potentially rescue decreased thymopoiesis and perturbed negative selection to significantly, albeit partially, restore defective central tolerance and reduce subclinical autoimmune symptoms in the elderly.
Conclusion
The thymus is an excellent and reasonably accessible target for rejuvenation with some potentially large payoffs for success. We have already seen the de-aging of the human thymus in a human trial back in 2019, and this study is yet another example of how thymic rejuvenation might be achieved in humans.
There are a number of possible approaches to the problem, some potentially better than others, and it really does look like a matter of time before one of them reaches humans. The way things are looking, it isn’t going to be too long before that happens and opens up the possibilities of healthy longevity through rejuvenation biotechnology.