Scientists have linked disrupted Ca²⁺ homeostasis to aging in both progeroid and naturally aging mice. Rescuing it with a well-known antidepressant significantly increased the animals’ median and maximum lifespan [1].
Calcium ions in progeria and natural aging
Cells use calcium ions (Ca²⁺) for signaling, and a rise or fall in cytoplasmic Ca²⁺ switches many pathways on or off. Ca²⁺ homeostasis is known to break down in many age-related diseases, including heart failure, hypertension, sarcopenia, and Alzheimer’s [2]. What has been missing is a concrete molecular chain that connects this phenomenon to cellular aging. A new study from Chinese scientists, published in Nature Communications, aims to bridge that gap.
Prior research had found that Ca²⁺ is elevated in cells taken from patients with Hutchinson-Gilford progeria syndrome (HGPS), a rare genetic disease characterized by accelerated aging [3]. However, nobody had previously shown that this Ca²⁺ disruption causes the aging phenotype or explained how.
The inflammation connection
The researchers started by applying unbiased proteomics on lung tissue from 12-week-old normal and progeroid mice. The Ca²⁺ pathway was among the most upregulated in the latter, and so was one of the proteins involved in it, S100A6. The team confirmed S100A6 elevation across heart, lung, skin, and muscle of progeroid mice and in fibroblasts from HGPS patients. Knocking down S100A6 in HGPS patient cells lowered the DNA damage marker γH2AX and the senescence markers p16 and p21, and it restored the cells’ ability to divide.
The authors then asked where the excess Ca²⁺ was coming from and found cytoplasmic Ca²⁺ elevated across multiple tissues and in patient cells, with a matching drop in the endoplasmic reticulum (ER), the cell’s main protein-producing organelle. Since mitochondrial Ca²⁺ stayed stable, it became obvious that the ER was the leak’s source. The leak channel turned out to be IP3R, the main Ca²⁺ release valve on the ER, which was overexpressed in patient cells. Blocking IP3R lowered both Ca²⁺ and cytoplasmic S100A6.
To find S100A6’s partners, the researchers used mass spectrometry and identified PARP1, a central DNA-repair enzyme, as a binding partner. PARP1 is known to be downregulated in HGPS. Knocking down S100A6 raised the PARP1 protein level without changing its messenger RNA (mRNA) source, meaning the regulation happens at the protein level: the same amount is produced, but less is degraded, as was confirmed by further experiments.
How does PARP1 loss accelerate aging? Cells overexpressing progerin, the main culprit in HGPS, showed increased signs of DNA breaks and more cytoplasmic chromatin fragments (CCF) – bits of DNA that escape the nucleus and trigger inflammation via the cGAS-STING pathway. Inflammation and DNA break markers were reduced by S100A6 knockdown, while inhibiting PARP1 and forcing CCF formation reversed the effect.
Mianserin improves healthspan and lifespan
Inhibiting IP3R improved body weight and locomotion, and it extended median survival by 14.15% in progeroid mice, but the drug used (2-APB) also caused tremors, which is a dealbreaker for long-term therapy. This pushed the researchers to look for a more tolerable option.
What they found was mianserin, a long-known anti-depression medication. It antagonizes serotonin receptors that normally activate IP3R, so blocking them should shut the Ca²⁺ leak from ER. Mianserin indeed reversed senescence markers, restored proliferation, and reduced γH2AX and CCF. Artificially raising intracellular Ca²⁺ abolished these anti-aging effects.
Treating progeroid mice with mianserin from 4 weeks of age extended median survival by about 30% and improved appearance, weight, cardiac, pulmonary, and muscle function. It also lowered S100A6 and inflammatory factors while raising PARP1 – closing the loop back to the proposed mechanism.
Confirming that S100A6 is relevant to normal aging, the researchers found that it was elevated in senescent fibroblasts, in skin fibroblasts from very old people, and in aged rat tissues. Interestingly, S100A6 was actually reduced in models where senescence was caused by acute stress, suggesting that high cytoplasmic S100A6 is a signature specifically of chronic/physiological senescence, not senescence in general.
Finally, the team treated naturally aged mice with mianserin or saline every other day for four months. Mianserin-treated mice had larger body size, glossier fur, less spinal curvature (kyphosis), less leg osteoporosis, slowed weight loss, and better locomotion. The treatment also extended median survival by 17.5%.
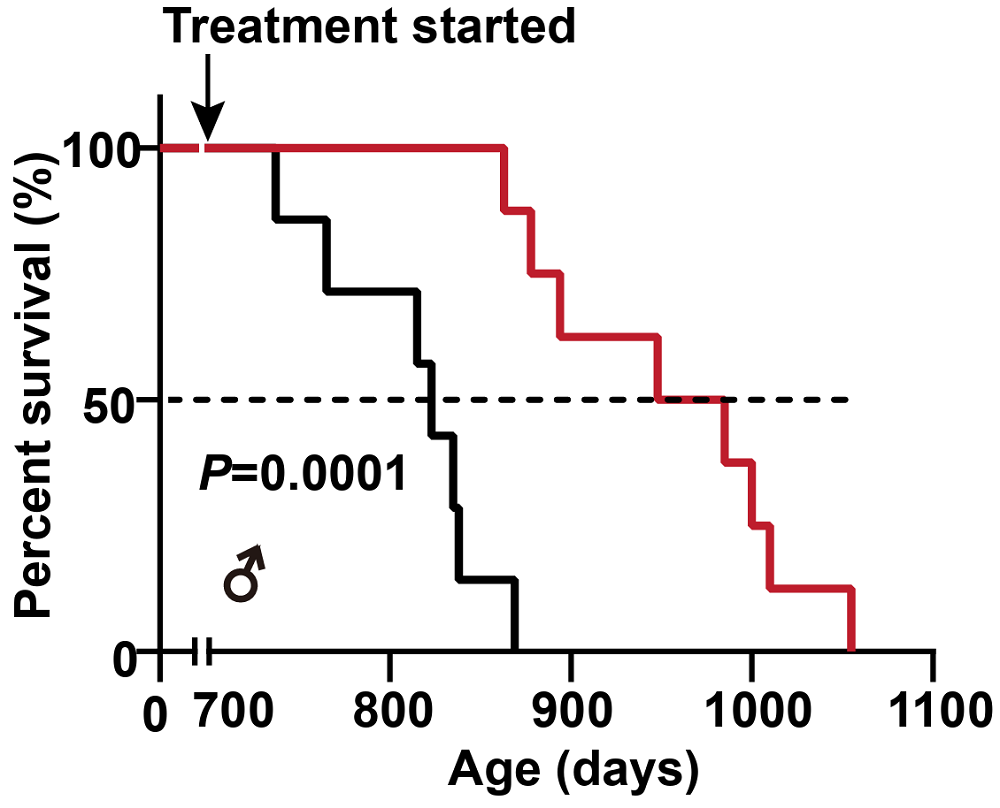
This result is quite impressive, especially considering how late in life the treatment began, but it comes with several important caveats. First, the size of this healthspan/lifespan cohort was small, with 7 or 8 mice in each group. Second, the controls’ lifespan was low for Black 6 mice (823 days median and 869 days maximum). This problem plagues many longevity studies [4], but here, healthspan experiments provide some reassurance by pointing in the same direction and suggesting that “real” slowing of aging is happening. Finally, the cohort was entirely male, which makes it harder to generalize results.
Literature
[1] Xiang, W., Hu, Q., Sun, P., Wu, X., Jiang, H., Qu, M., … & Zhang, Y. (2026). Ameliorating calcium homeostasis improves longevity and healthspan in progeroid and naturally aged mice. Nature Communications.
[2] Berridge, M. J. (2012). Calcium signalling remodelling and disease. Biochemical Society Transactions, 40(2), 297-309.
[3] Fafián-Labora, J. A., Morente-López, M., de Toro, F. J., & Arufe, M. C. (2021). High-throughput screen detects calcium signaling dysfunction in hutchinson-gilford progeria syndrome. International journal of molecular sciences, 22(14), 7327.
[4] Pabis, K., Barardo, D., Gruber, J., Sirbu, O., Malavolta, M., Selvarajoo, K., … & Kennedy, B. K. (2024). The impact of short-lived controls on the interpretation of lifespan experiments and progress in geroscience–Through the lens of the “900-day rule”. Ageing Research Reviews, 101, 102512.